
Live-Cell Analysis of Human Cytomegalovirus DNA Polymerase Holoenzyme Assembly by Resonance Energy Transfer Methods




Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture
2.3. Western Blotting Analysis
2.4. Indirect Immunofluorescence Analysis
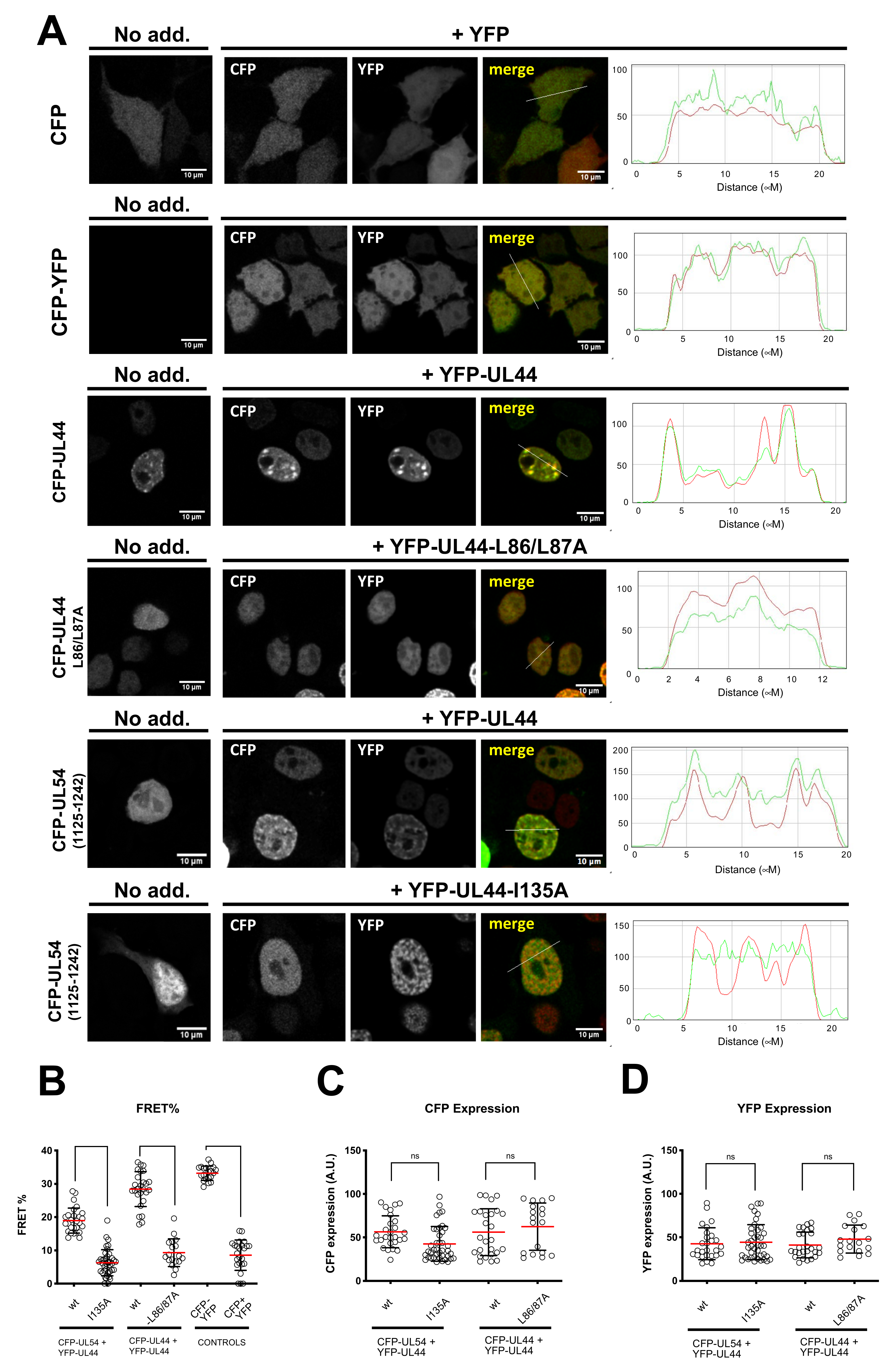
2.5. FRET Acceptor Photobleaching Assays
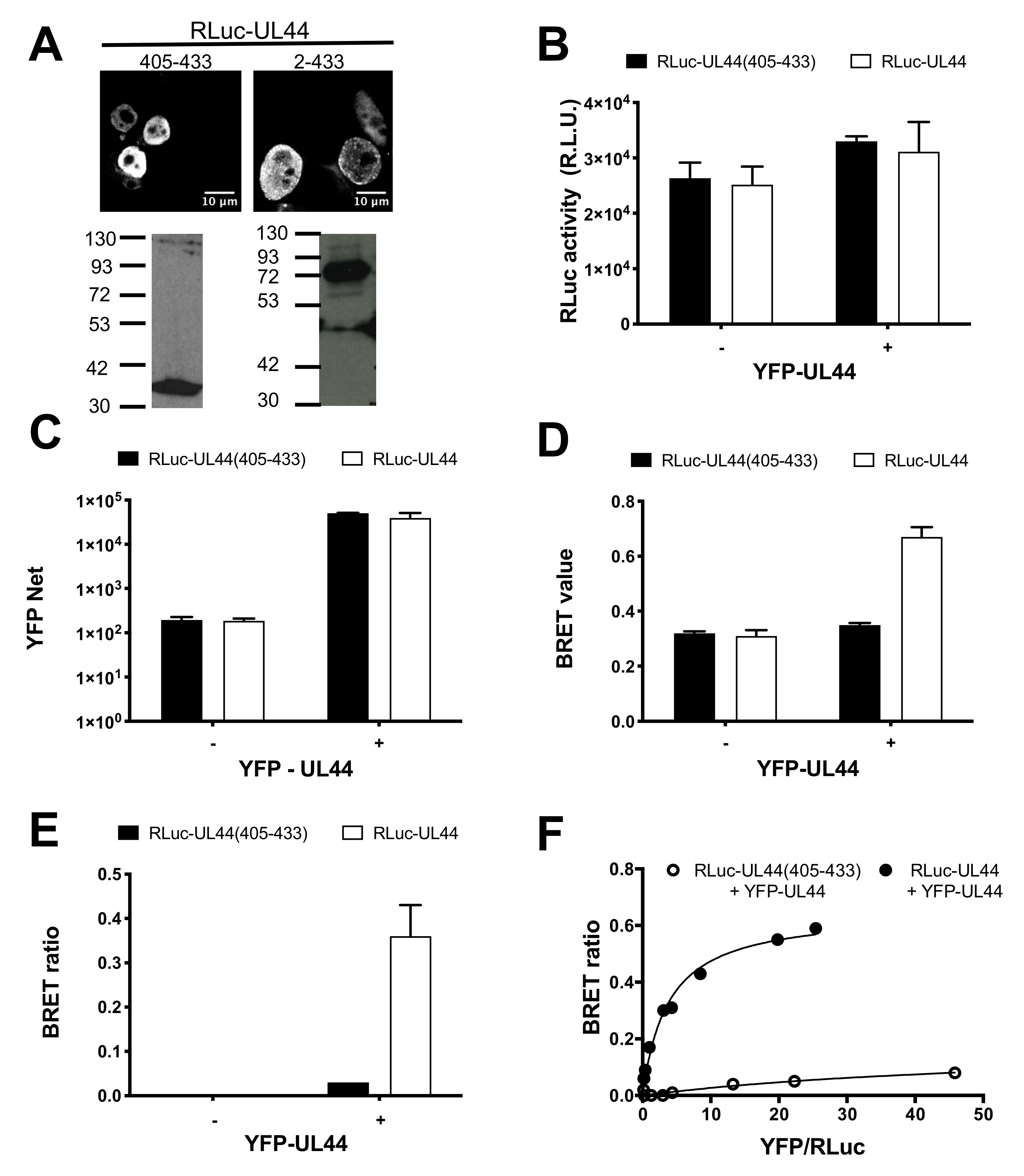
2.6. BRET Assays
(RLuc emission) BRET donor
2.7. Generation of Lentiviral Particles
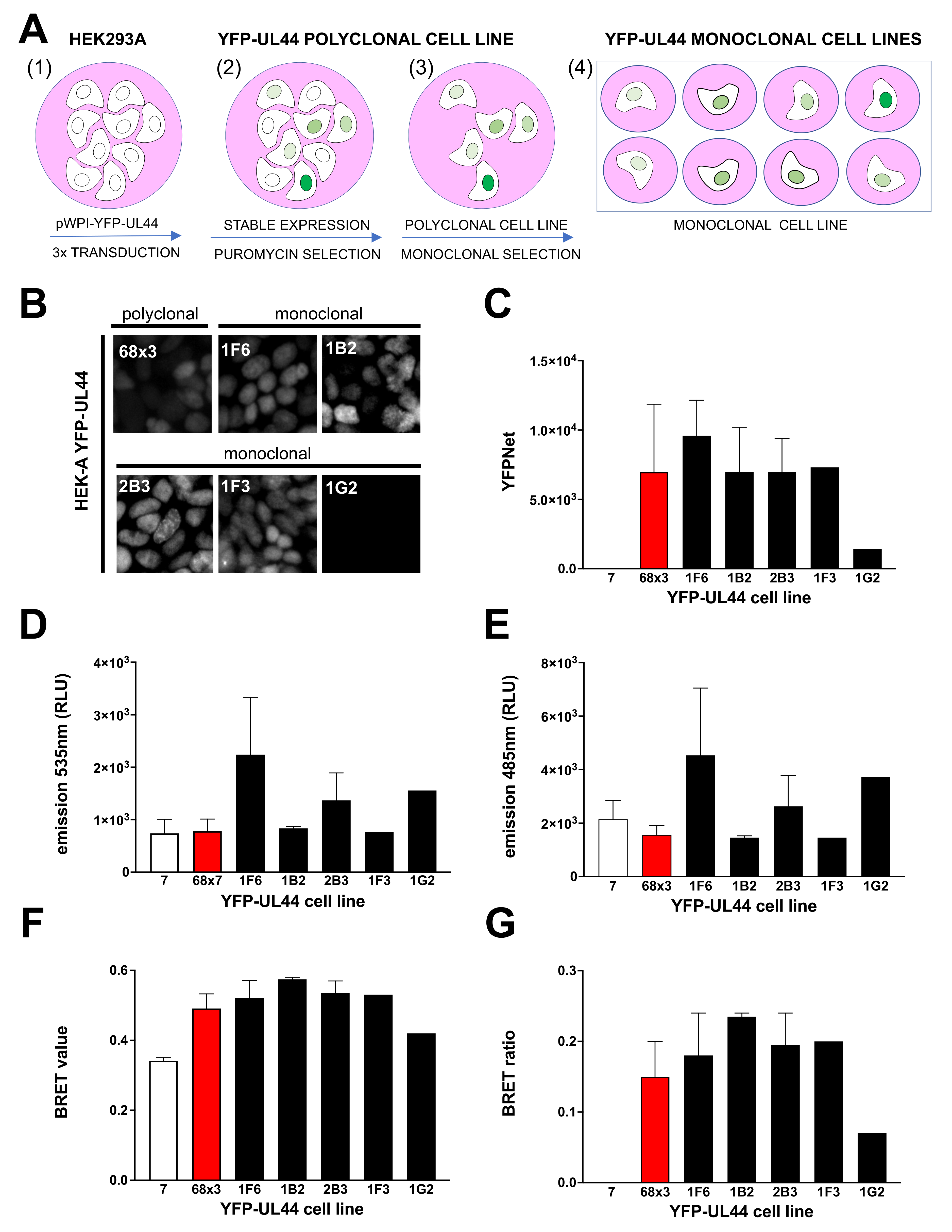
2.8. Generation of Polyclonal and Monoclonal HEK293A Cells Stably Expressing YFP-UL44
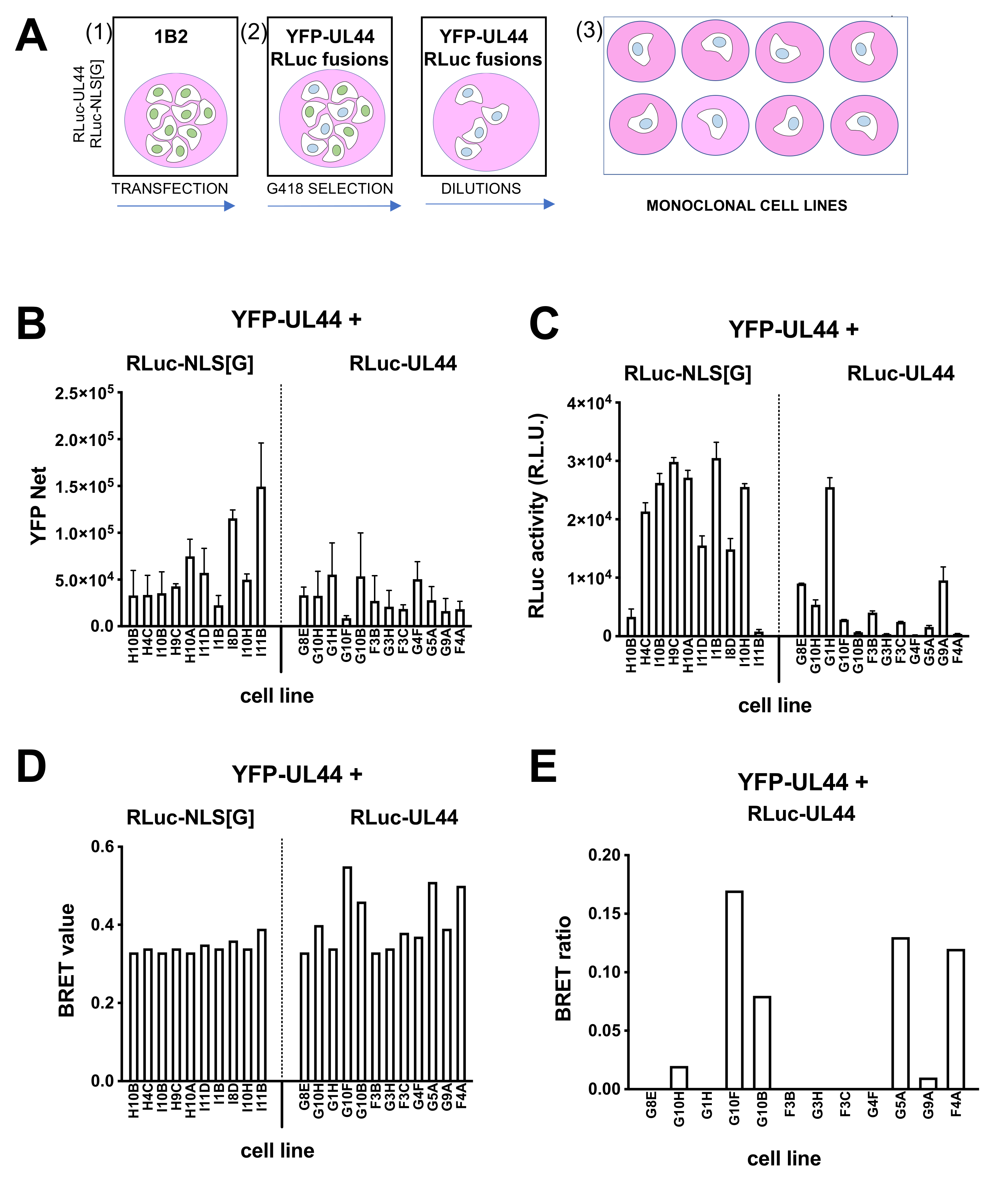
2.9. Generation of Polyclonal and Monoclonal HEK293A Cells Stably Expressing YFP-UL44 and Either Rluc-NLS[G] or Rluc-UL44
2.10. Statistical Analysis
3. Results
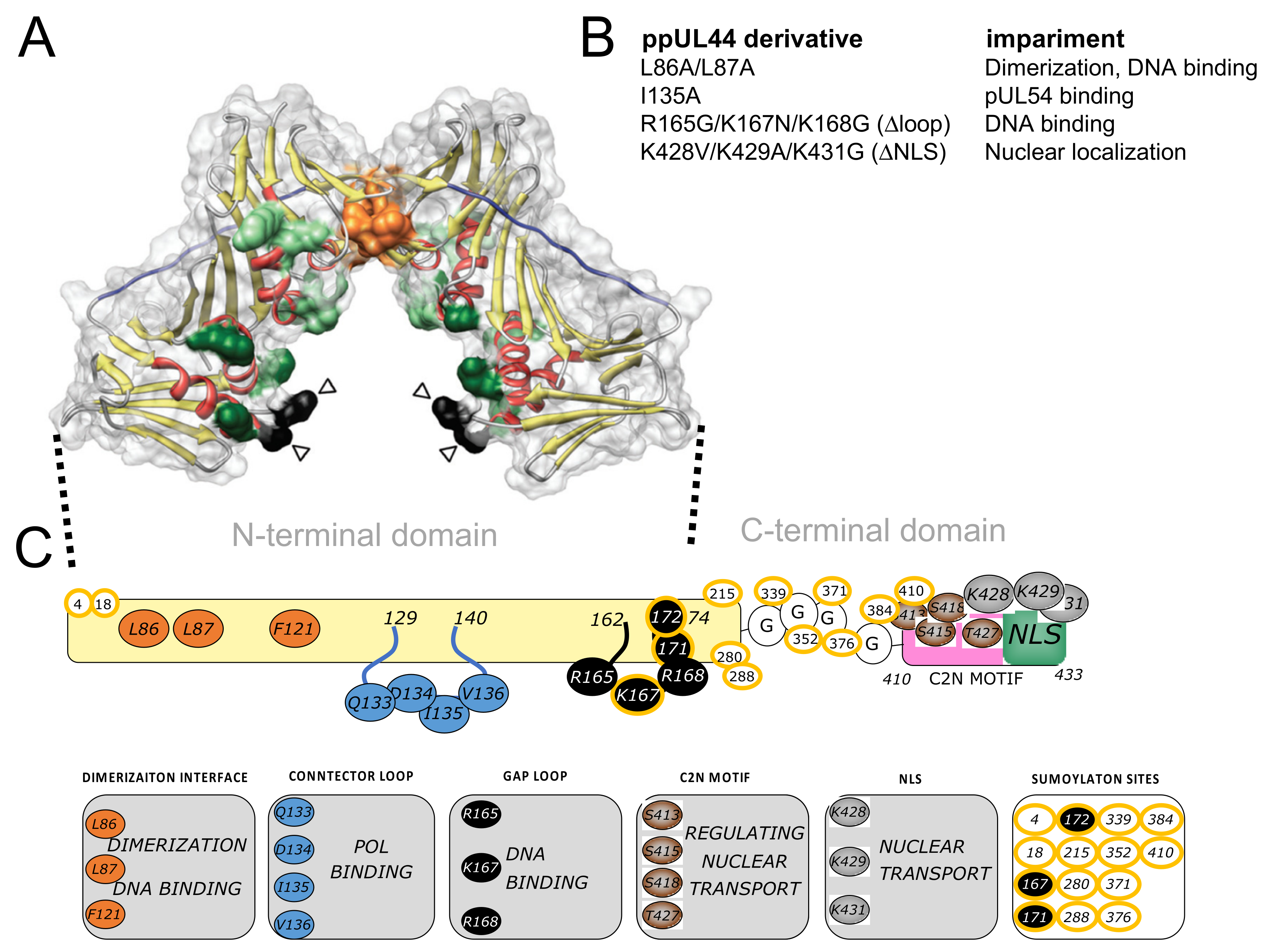
3.1. ppUL44 Residues Leucine 86/87 and Isoleucine 135 Are Crucial for Its Dimerization and Interaction with pUL54 in Cells, Respectively
3.2. BRET Assays Allow to Quantify ppUL44 Homodimerization and Holoenzyme Formation in Living Cells
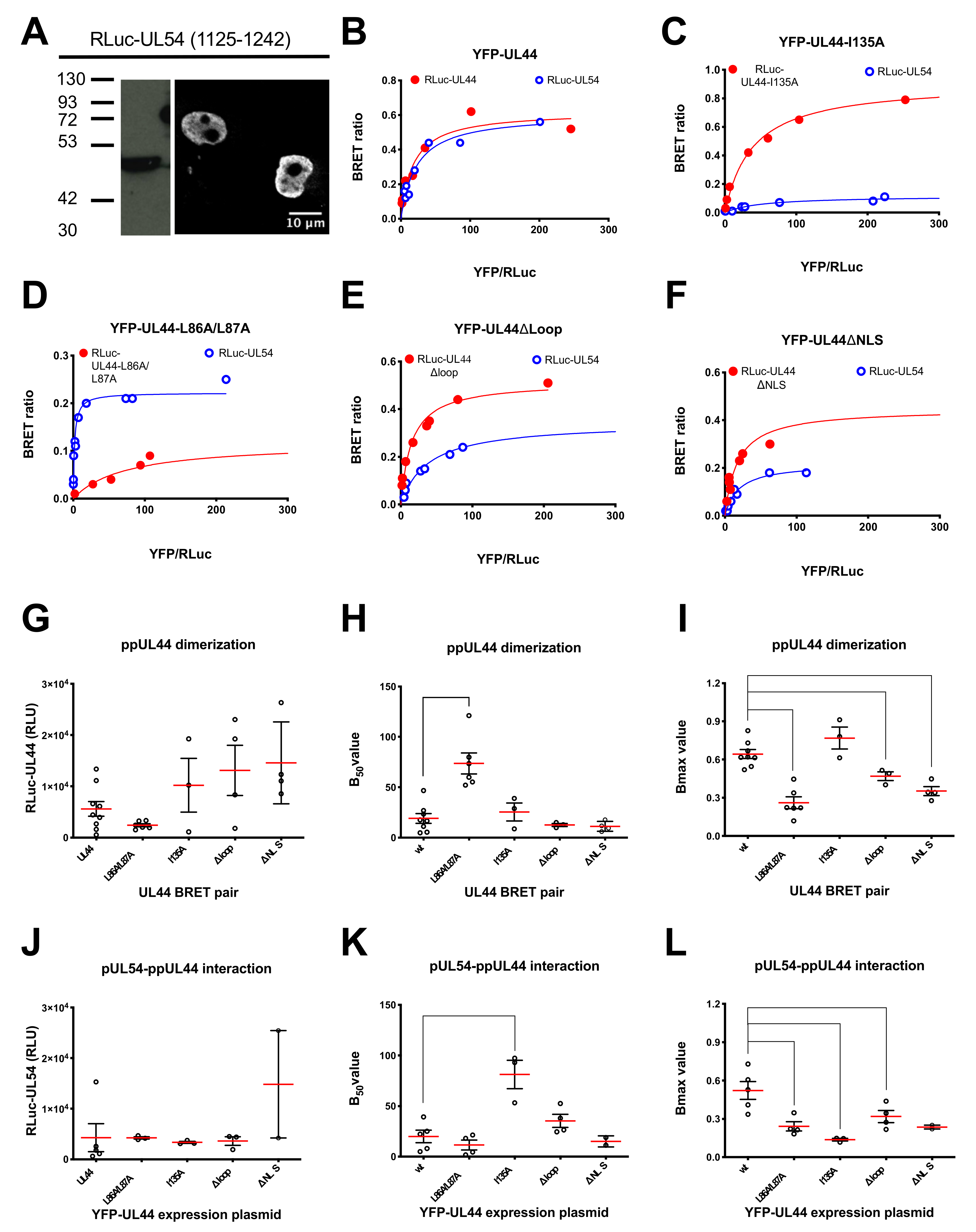
3.3. ppUL44 Dimerizes in Cells with an Affinity Comparable to That of the ppUL44-pUL54 Interaction
3.4. Determination of the Impact of Specific Amino Acids Substitutions within ppUL44 Functional Domains on Protein Homodimerization and DNA Polymerase Holoenzyme Formation
3.5. Generation of YFP-UL44 Stable Cell Lines
3.6. Generation of YFP-UL44/RLuc-UL44 Stable Cell Lines
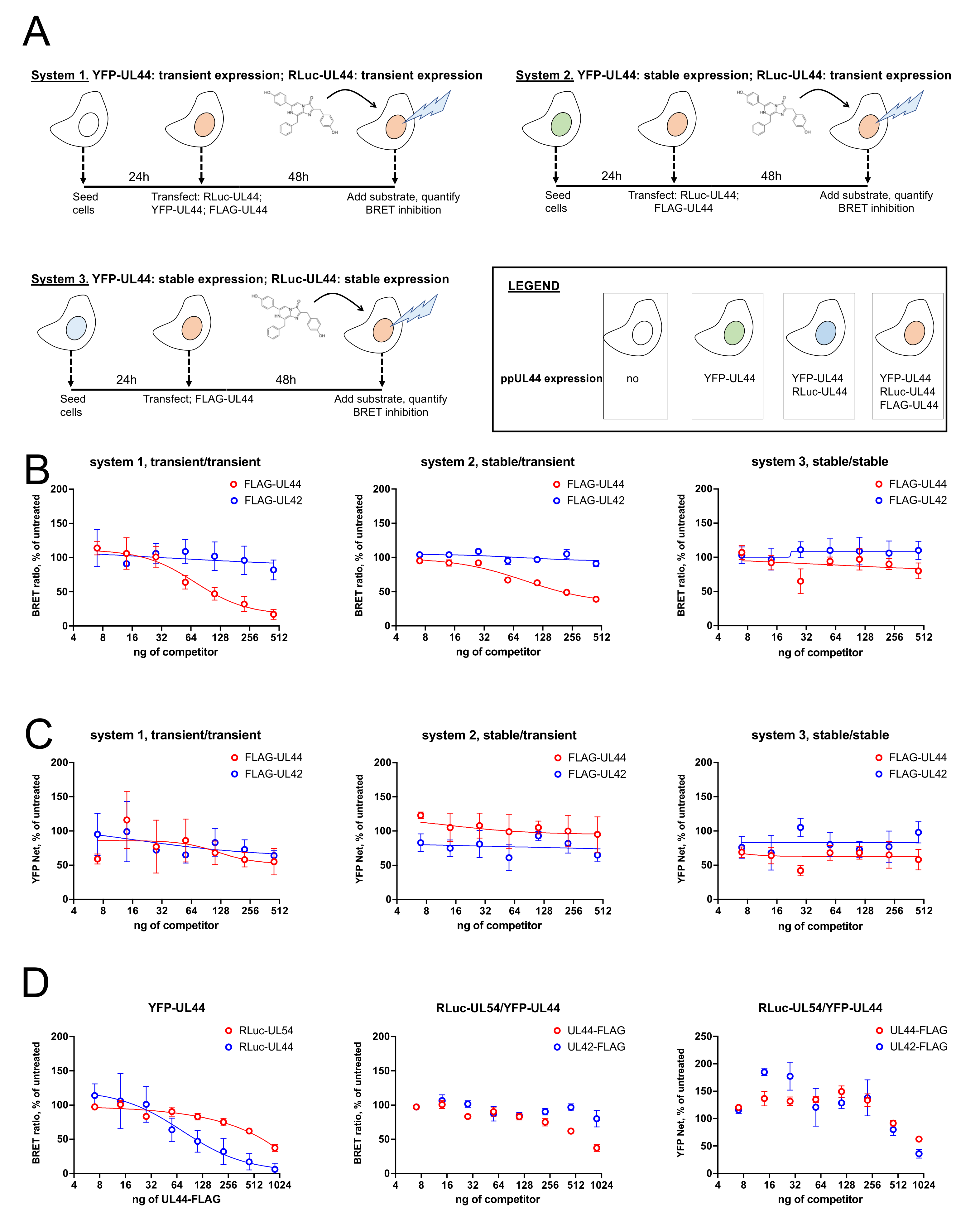
3.7. Transient Expression of RLuc-UL44 Allows to Monitor Interference with RLuc-UL44/YFP-UL44 and RLuc-UL54/YFP-UL44 Interactions upon Overexpression of FLAG-UL44
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvisi, G.; Jans, D.A.; Camozzi, D.; Avanzi, S.; Loregian, A.; Ripalti, A.; Palu, G. Regulated transport into the nucleus of herpesviridae DNA replication core proteins. Viruses 2013, 5, 2210–2234. [Google Scholar] [CrossRef] [PubMed]
- Pari, G.S. Nuts and bolts of human cytomegalovirus lytic DNA replication. Curr. Top. Microbiol. Immunol. 2008, 325, 153–166. [Google Scholar] [PubMed]
- Pari, G.S.; Anders, D.G. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 1993, 67, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Appleton, B.A.; Loregian, A.; Filman, D.J.; Coen, D.M.; Hogle, J.M. The cytomegalovirus DNA polymerase subunit UL44 forms a C clamp-shaped dimer. Mol. Cell 2004, 15, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Murayama, K.; Nakayama, S.; Kato-Murayama, M.; Akasaka, R.; Ohbayashi, N.; Kamewari-Hayami, Y.; Terada, T.; Shirouzu, M.; Tsurumi, T.; Yokoyama, S. Crystal structure of epstein-barr virus DNA polymerase processivity factor BMRF1. J. Biol. Chem. 2009, 284, 35896–35905. [Google Scholar] [CrossRef]
- Zuccola, H.J.; Filman, D.J.; Coen, D.M.; Hogle, J.M. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 2000, 5, 267–278. [Google Scholar] [CrossRef]
- Baltz, J.L.; Filman, D.J.; Ciustea, M.; Silverman, J.E.; Lautenschlager, C.L.; Coen, D.M.; Ricciardi, R.P.; Hogle, J.M. The crystal structure of PF-8, the DNA polymerase accessory subunit from Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2009, 83, 12215–12228. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Loregian, A.; Appleton, B.A.; Hogle, J.M.; Coen, D.M. Specific residues in the connector loop of the human cytomegalovirus DNA polymerase accessory protein UL44 are crucial for interaction with the UL54 catalytic subunit. J. Virol. 2004, 78, 9084–9092. [Google Scholar] [CrossRef]
- Randell, J.C.W.; Komazin, G.; Jiang, C.; Hwang, C.B.C.; Coen, D.M. Effects of substitutions of arginine residues on the basic surface of herpes simplex virus UL42 support a role for DNA binding in processive DNA synthesis. J. Virol. 2005, 79, 12025–12034. [Google Scholar] [CrossRef]
- Komazin-Meredith, G.; Santos, W.L.; Filman, D.J.; Hogle, J.M.; Verdine, G.L.; Coen, D.M. The positively charged surface of herpes simplex virus UL42 mediates DNA binding. J. Biol. Chem. 2008, 283, 6154–6161. [Google Scholar] [CrossRef] [PubMed]
- Weiland, K.L.; Oien, N.L.; Homa, F.; Wathen, M.W. Functional analysis of human cytomegalovirus polymerase accessory protein. Virus Res. 1994, 34, 191–206. [Google Scholar] [CrossRef]
- Alvisi, G.; Jans, D.A.; Ripalti, A. Human cytomegalovirus (HCMV) DNA polymerase processivity factor ppUL44 dimerizes in the cytosol before translocation to the nucleus. Biochemistry 2006, 45, 6866–6872. [Google Scholar] [CrossRef] [PubMed]
- Appleton, B.A.; Brooks, J.; Loregian, A.; Filman, D.J.; Coen, D.M.; Hogle, J.M. Crystal structure of the cytomegalovirus DNA polymerase subunit UL44 in complex with the C terminus from the catalytic subunit. Differences in structure and function relative to unliganded UL44. J. Biol. Chem. 2006, 281, 5224–5232. [Google Scholar] [CrossRef]
- Gallo, M.L.; Dorsky, D.I.; Crumpacker, C.S.; Parris, D.S. The essential 65-kilodalton DNA-binding protein of herpes simplex virus stimulates the virus-encoded DNA polymerase. J. Virol. 1989, 63, 5023–5029. [Google Scholar] [CrossRef]
- Gibson, W.; Murphy, T.L.; Roby, C. Cytomegalovirus-infected cells contain a DNA-binding protein. Virology 1981, 111, 251–262. [Google Scholar] [CrossRef]
- Ripalti, A.; Boccuni, M.C.; Campanini, F.; Landini, M.P. Cytomegalovirus-mediated induction of antisense mRNA expression to UL44 inhibits virus replication in an astrocytoma cell line: Identification of an essential gene. J. Virol. 1995, 69, 2047–2057. [Google Scholar] [CrossRef]
- Alvisi, G.; Jans, D.A.; Guo, J.; Pinna, L.A.; Ripalti, A. A protein kinase CK2 site flanking the nuclear targeting signal enhances nuclear transport of human cytomegalovirus ppUL44. Traffic 2005, 6, 1002–1013. [Google Scholar] [CrossRef]
- Alvisi, G.; Marin, O.; Pari, G.; Mancini, M.; Avanzi, S.; Loregian, A.; Jans, D.A.; Ripalti, A. Multiple phosphorylation sites at the C-terminus regulate nuclear import of HCMV DNA polymerase processivity factor ppUL44. Virology 2011, 417, 259–267. [Google Scholar] [CrossRef]
- Boccuni, M.C.; Campanini, F.; Battista, M.C.; Bergamini, G.; Dal Monte, P.; Ripalti, A.; Landini, M.P. Human cytomegalovirus product UL44 downregulates the transactivation of HIV-1 long terminal repeat. AIDS 1998, 12, 365–372. [Google Scholar] [CrossRef]
- Silva, L.A.; Loregian, A.; Pari, G.S.; Strang, B.L.; Coen, D.M. The carboxy-terminal segment of the human cytomegalovirus DNA polymerase accessory subunit UL44 is crucial for viral replication. J. Virol. 2010, 84, 11563–11568. [Google Scholar] [CrossRef]
- Silva, L.A.; Strang, B.L.; Lin, E.W.; Kamil, J.P.; Coen, D.M. Sites and roles of phosphorylation of the human cytomegalovirus DNA polymerase subunit UL44. Virology 2011, 417, 268–280. [Google Scholar] [CrossRef][Green Version]
- Sinigalia, E.; Alvisi, G.; Mercorelli, B.; Coen, D.M.; Pari, G.S.; Jans, D.A.; Ripalti, A.; Palu, G.; Loregian, A. Role of homodimerization of human cytomegalovirus DNA polymerase accessory protein UL44 in origin-dependent DNA replication in cells. J. Virol. 2008, 82, 12574–12579. [Google Scholar] [CrossRef]
- Komazin-Meredith, G.; Petrella, R.J.; Santos, W.L.; Filman, D.J.; Hogle, J.M.; Verdine, G.L.; Karplus, M.; Coen, D.M. The human cytomegalovirus UL44 C clamp wraps around DNA. Structure 2008, 16, 1214–1225. [Google Scholar] [CrossRef]
- Jiang, C.Y.; Hwang, Y.T.; Randell, J.C.W.; Coen, D.M.; Hwang, C.B.C. Mutations that decrease DNA binding of the processivity factor of the herpes simplex virus DNA polymerase reduce viral yield, alter the kinetics of viral DNA replication, and decrease the fidelity of DNA replication. J. Virol. 2007, 81, 3495–3502. [Google Scholar] [CrossRef]
- Alvisi, G.; Roth, D.M.; Camozzi, D.; Pari, G.S.; Loregian, A.; Ripalti, A.; Jans, D.A. The flexible loop of the human cytomegalovirus DNA polymerase processivity factor ppUL44 is required for efficient DNA binding and replication in cells. J. Virol. 2009, 83, 9567–9576. [Google Scholar] [CrossRef]
- Chen, H.; Coseno, M.; Ficarro, S.B.; Mansueto, M.S.; Komazin-Meredith, G.; Boissel, S.; Filman, D.J.; Marto, J.A.; Hogle, J.M.; Coen, D.M. A Small Covalent Allosteric Inhibitor of Human Cytomegalovirus DNA Polymerase Subunit Interactions. ACS Infect. Dis. 2017, 3, 112–118. [Google Scholar] [CrossRef]
- Loregian, A.; Rigatti, R.; Murphy, M.; Schievano, E.; Palu, G.; Marsden, H.S. Inhibition of human cytomegalovirus DNA polymerase by C-terminal peptides from the UL54 subunit. J. Virol. 2003, 77, 8336–8344. [Google Scholar] [CrossRef]
- Loregian, A.; Coen, D.M. Selective anti-cytomegalovirus compounds discovered by screening for inhibitors of subunit interactions of the viral polymerase. Chem. Biol. 2006, 13, 191–200. [Google Scholar] [CrossRef]
- Smith, K.M.; Tsimbalyuk, S.; Edwards, M.R.; Cross, E.M.; Batra, J.; Soares da Costa, T.P.; Aragao, D.; Basler, C.F.; Forwood, J.K. Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat. Commun. 2018, 9, 3703. [Google Scholar] [CrossRef]
- Trevisan, M.; Di Antonio, V.; Radeghieri, A.; Palu, G.; Ghildyal, R.; Alvisi, G. Molecular Requirements for Self-Interaction of the Respiratory Syncytial Virus Matrix Protein in Living Mammalian Cells. Viruses 2018, 10, 109. [Google Scholar] [CrossRef]
- Alvisi, G.; Paolini, L.; Contarini, A.; Zambarda, C.; Di Antonio, V.; Colosini, A.; Mercandelli, N.; Timmoneri, M.; Palu, G.; Caimi, L.; et al. Intersectin goes nuclear: Secret life of an endocytic protein. Biochem. J. 2018, 475, 1455–1472. [Google Scholar] [CrossRef]
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011, 85, 6963–6976. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Avanzi, S.; Musiani, D.; Camozzi, D.; Leoni, V.; Ly-Huynh, J.D.; Ripalti, A. Nuclear import of HSV-1 DNA polymerase processivity factor UL42 is mediated by a C-terminally located bipartite nuclear localization signal. Biochemistry 2008, 47, 13764–13777. [Google Scholar] [CrossRef]
- Elbadawy, H.M.; Mohammed Abdul, M.I.; Aljuhani, N.; Vitiello, A.; Ciccarese, F.; Shaker, M.A.; Eltahir, H.M.; Palu, G.; Di Antonio, V.; Ghassabian, H.; et al. Generation of Combinatorial Lentiviral Vectors Expressing Multiple Anti-Hepatitis C Virus shRNAs and Their Validation on a Novel HCV Replicon Double Reporter Cell Line. Viruses 2020, 12, 1044. [Google Scholar] [CrossRef]
- Smith, K.M.; Di Antonio, V.; Bellucci, L.; Thomas, D.R.; Caporuscio, F.; Ciccarese, F.; Ghassabian, H.; Wagstaff, K.M.; Forwood, J.K.; Jans, D.A.; et al. Contribution of the residue at position 4 within classical nuclear localization signals to modulating interaction with importins and nuclear targeting. Biochim. Biophys. Acta 2018, 1865, 1114–1129. [Google Scholar] [CrossRef]
- Scaturro, P.; Trist, I.M.; Paul, D.; Kumar, A.; Acosta, E.G.; Byrd, C.M.; Jordan, R.; Brancale, A.; Bartenschlager, R. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J. Virol. 2014, 88, 11540–11555. [Google Scholar] [CrossRef]
- Montagner, M.; Martello, G.; Piccolo, S. Monitoring Smad Activity In Vivo Using the Xenopus Model System. Methods Mol. Biol. 2016, 1344, 245–259. [Google Scholar]
- Sun, S.; Yang, X.; Wang, Y.; Shen, X. In Vivo Analysis of Protein-Protein Interactions with Bioluminescence Resonance Energy Transfer (BRET): Progress and Prospects. Int. J. Mol. Sci. 2016, 17, 1704. [Google Scholar] [CrossRef]
- Eberle, C.A.; Zayas, M.; Stukalov, A.; Pichlmair, A.; Alvisi, G.; Muller, A.C.; Bennett, K.L.; Bartenschlager, R.; Superti-Furga, G. The lysine methyltransferase SMYD3 interacts with hepatitis C virus NS5A and is a negative regulator of viral particle production. Virology 2014, 462–463, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Ripalti, A.; Ngankeu, A.; Giannandrea, M.; Caraffi, S.G.; Dias, M.M.; Jans, D.A. Human cytomegalovirus DNA polymerase catalytic subunit pUL54 possesses independently acting nuclear localization and ppUL44 binding motifs. Traffic 2006, 7, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Poddar, S.; Chakravarty, D.; Chakrabarti, P. Structural changes in DNA-binding proteins on complexation. Nucleic Acids Res. 2018, 46, 3298–3308. [Google Scholar] [CrossRef] [PubMed]
- Maga, G.; Hubscher, U. Proliferating cell nuclear antigen (PCNA): A dancer with many partners. J. Cell Sci. 2003, 116 Pt 15, 3051–3060. [Google Scholar] [CrossRef]
- Couturier, C.; Deprez, B. Setting Up a Bioluminescence Resonance Energy Transfer High throughput Screening Assay to Search for Protein/Protein Interaction Inhibitors in Mammalian Cells. Front. Endocrinol. 2012, 3, 100. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UL44 Derivative | RLuc-UL44 + YFP-UL44 | RLuc-UL54 + YFP-UL44 | ||
|---|---|---|---|---|
| Bmax | B50 | Bmax | B50 | |
| Wild-type | 0.64 ± 0.09 (8) | 19.0 ± 13.7 (8) | 0.52 ± 19.56 (5) | 20.0 ± 13.9 (5) |
| L86A/L87A 1,2,3 | 0.26 ± 0.11 (6) | 73.7 ± 25.7 (6) | 0.24 ± 0.07 (4) | 11.5 ± 9.8 (4) |
| I135A 3,4 | 0.77 ± 0.15 (3) | 25.5 ± 15.4 (3) | 0.14 ± 0.02 (3) | 81.2 ± 24.3 (3) |
| Δloop 2,3 | 0.47 ± 0.06 (3) | 12.6 ± 2.6 (3) | 0.32 ± 0.10 (4) | 34.4 ± 13.0 (4) |
| ΔNLS 3,5 | 0.35 ± 0.07 (4) | 11.2 ± 4.9(4) | 0.24 ± 0.02 (2) | 15.1 ± 5.4 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Antonio, V.; Palù, G.; Alvisi, G. Live-Cell Analysis of Human Cytomegalovirus DNA Polymerase Holoenzyme Assembly by Resonance Energy Transfer Methods. Microorganisms 2021, 9, 928. https://doi.org/10.3390/microorganisms9050928
Di Antonio V, Palù G, Alvisi G. Live-Cell Analysis of Human Cytomegalovirus DNA Polymerase Holoenzyme Assembly by Resonance Energy Transfer Methods. Microorganisms. 2021; 9(5):928. https://doi.org/10.3390/microorganisms9050928
Chicago/Turabian StyleDi Antonio, Veronica, Giorgio Palù, and Gualtiero Alvisi. 2021. "Live-Cell Analysis of Human Cytomegalovirus DNA Polymerase Holoenzyme Assembly by Resonance Energy Transfer Methods" Microorganisms 9, no. 5: 928. https://doi.org/10.3390/microorganisms9050928
APA StyleDi Antonio, V., Palù, G., & Alvisi, G. (2021). Live-Cell Analysis of Human Cytomegalovirus DNA Polymerase Holoenzyme Assembly by Resonance Energy Transfer Methods. Microorganisms, 9(5), 928. https://doi.org/10.3390/microorganisms9050928