
Comparing Sediment Microbiomes in Contaminated and Pristine Wetlands along the Coast of Yucatan

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Processing
2.2. DNA Extraction and Sequencing
2.3. Data Analysis
3. Results
3.1. Taxonomic Composition of Prokaryotic Communities of the Yucatan Coast
3.2. Diversity Estimates
3.3. Environment–Taxonomy Association
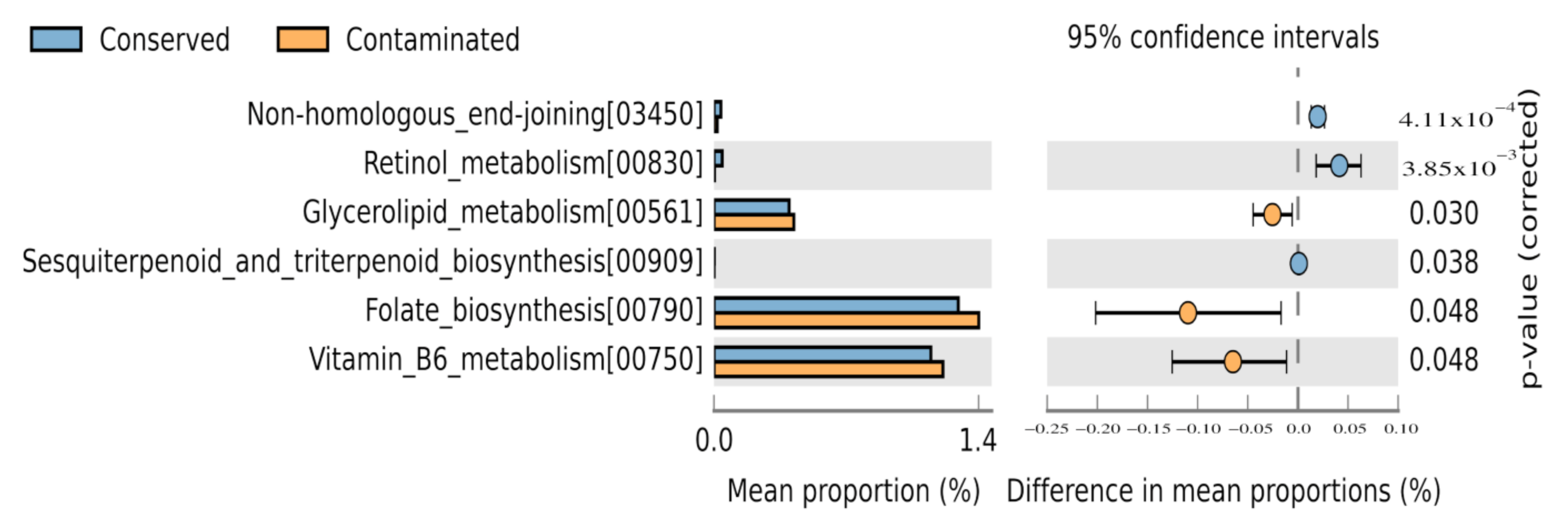
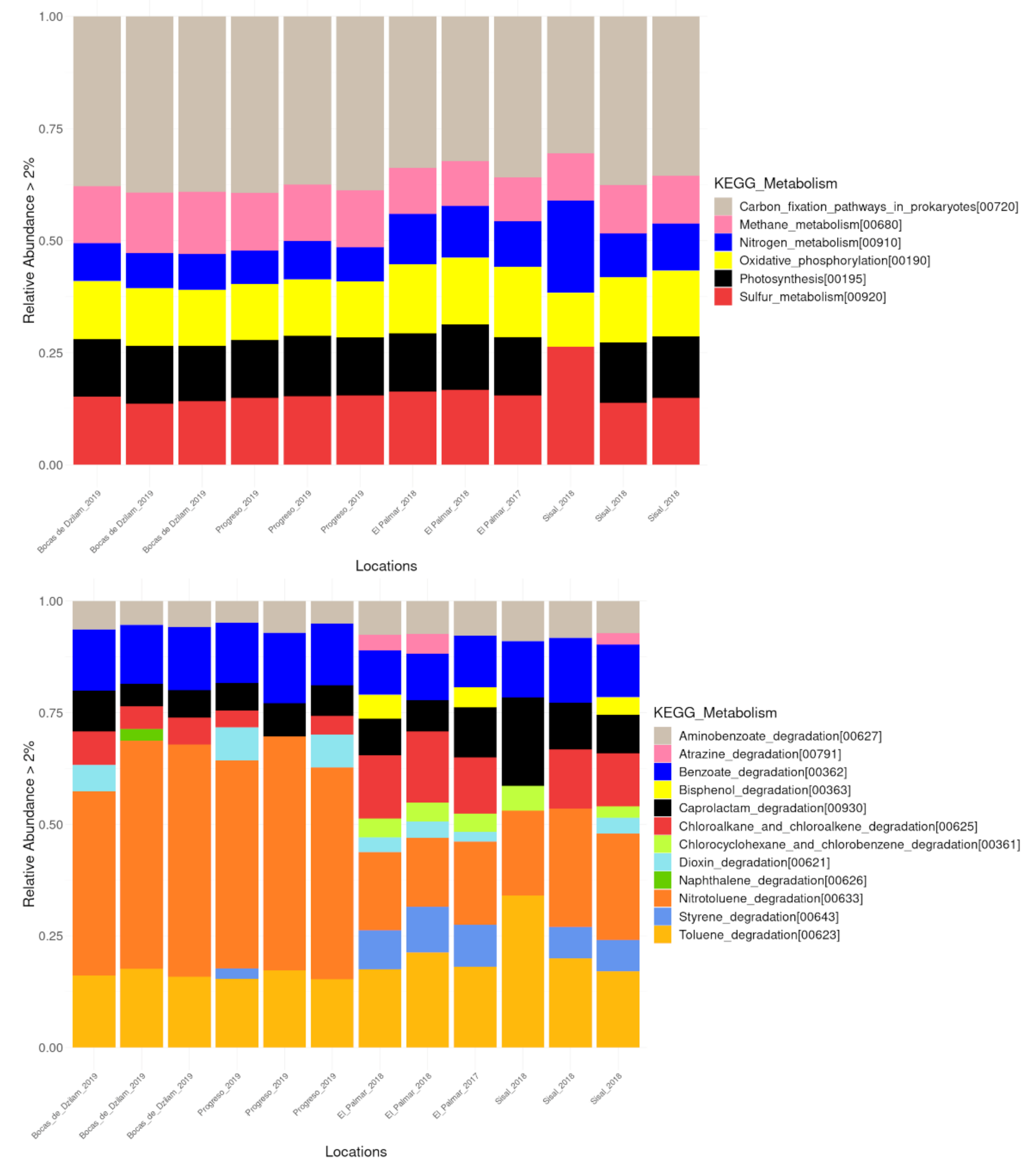
3.4. Functional Profile Prediction
3.5. Environmental Metabolic Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quero, G.M.; Cassin, D.; Botter, M.; Perini, L.; Luna, G.M. Patterns of benthic bacterial diversity in coastal areas contaminated by heavy metals, polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs). Front. Microbiol. 2015, 6, 1053. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.Y.; Dafforn, K.A.; Brown, M.V.; Johnston, E.L. Bacterial communities are sensitive indicators of contaminant stress. Mar. Pollut. Bull. 2012, 64, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Lazar, C.S.; Teske, A.P.; Dick, G.J. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 2015, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheng, H.-F.; He, Y.; Wu, J.-Y.; Jiang, Y.-X.; Tam, N.F.-Y.; Zhou, H.-W. Comparison of the Levels of Bacterial Diversity in Freshwater, Intertidal Wetland, and Marine Sediments by Using Millions of Illumina Tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef]
- Choi, H.; Koh, H.-W.; Kim, H.; Chae, J.-C.; Park, S.-J. Microbial Community Composition in the Marine Sediments of Jeju Island: Next-Generation Sequencing Surveys. J. Microbiol. Biotechnol. 2016, 26, 883–890. [Google Scholar] [CrossRef]
- Labbate, M.; Seymour, J.R.; Lauro, F.; Brown, M.V. Anthropogenic Impacts on the Microbial Ecology and Function of Aquatic Environments. Front. Microbiol. 2016, 7, 1044. [Google Scholar] [CrossRef]
- Bauer-Gottwein, P.; Gondwe, B.R.N.; Charvet, G.; Marin, L.E.; Rebolledo-Vieyra, M.; Merediz-Alonso, G. The Yucatán Peninsula karst aquifer, Mexico. Hydrogeol. J. 2011, 19, 507–524. [Google Scholar] [CrossRef]
- Suárez-Moo, P.; Lamelas, A.; Garcia-Bautista, I.; Barahona-Pérez, L.F.; Sandoval-Flores, G.; Valdes-Lozano, D.; Toledano-Thompson, T.; Polanco-Lugo, E.; Valdez-Ojeda, R. Characterization of sediment microbial communities at two sites with low hydrocarbon pollution in the southeast Gulf of Mexico. PeerJ 2020, 8, e10339. [Google Scholar] [CrossRef]
- Javan, G.T.; Finley, S.J.; Smith, T.; Miller, J.; Wilkinson, J.E. Cadaver Thanatomicrobiome Signatures: The Ubiquitous Nature of Clostridium Species in Human Decomposition. Front. Microbiol. 2017, 8, 2096. [Google Scholar] [CrossRef]
- Belikov, S.; Belkova, N.; Butina, T.; Chernogor, L.; Kley, A.M.-V.; Nalian, A.; Rorex, C.; Khanaev, I.; Maikova, O.; Feranchuk, S. Diversity and shifts of the bacterial community associated with Baikal sponge mass mortalities. PLoS ONE 2019, 14, e0213926. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference. BioRxiv 2019. preprint. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Ullah, R.; Yasir, M.; Khan, I.; Bibi, F.; Sohrab, S.S.; Al-Ansari, A.; Al-Abbasi, F.; Al-Sofyani, A.A.; Daur, I.; Lee, S.-W.; et al. Comparative bacterial community analysis in relatively pristine and anthropogenically influenced mangrove ecosystems on the Red Sea. Can. J. Microbiol. 2017, 63, 649–660. [Google Scholar] [CrossRef]
- Jiang, X.-T.; Peng, X.; Deng, G.-H.; Sheng, H.-F.; Wang, Y.; Zhou, H.-W.; Tam, N.F.-Y. Illumina Sequencing of 16S rRNA Tag Revealed Spatial Variations of Bacterial Communities in a Mangrove Wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef]
- Haldar, S.; Nazareth, S.W. Taxonomic diversity of bacteria from mangrove sediments of Goa: Metagenomic and functional analysis. 3 Biotech 2018, 8, 436. [Google Scholar] [CrossRef]
- Wu, P.; Xiong, X.; Xu, Z.; Lu, C.; Cheng, H.; Lyu, X.; Zhang, J.; He, W.; Deng, W.; Lyu, Y.; et al. Bacterial Communities in the Rhizospheres of Three Mangrove Tree Species from Beilun Estuary, China. PLoS ONE 2016, 11, e0164082. [Google Scholar] [CrossRef]
- Xu, Y.; He, Y.; Egidi, E.; Franks, A.E.; Tang, C.; Xu, J. Pentachlorophenol alters the acetate-assimilating microbial community and redox cycling in anoxic soils. Soil Biol. Biochem. 2019, 131, 133–140. [Google Scholar] [CrossRef]
- Tahon, G.; Tytgat, B.; Lebbe, L.; Carlier, A.; Willems, A. Abditibacterium utsteinense sp. nov., the first cultivated member of candidate phylum FBP, isolated from ice-free Antarctic soil samples. Syst. Appl. Microbiol. 2018, 41, 279–290. [Google Scholar] [CrossRef]
- Ziganshina, E.E.; Ibragimov, E.M.; Ilinskaya, O.N.; Ziganshin, A.M. Bacterial communities inhabiting toxic industrial wastewater generated during nitrocellulose production. Biologia 2016, 71, 70–78. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Garcia, R.; Müller, R. The Family Haliangiaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 173–181. [Google Scholar]
- Farag, I.F.; Youssef, N.H.; Elshahed, M.S. Global Distribution Patterns and Pangenomic Diversity of the Candidate Phylum “Latescibacteria” (WS3). Appl. Environ. Microbiol. 2017, 83, e00521-17. [Google Scholar] [CrossRef]
- Youssef, N.H.; Farag, I.F.; Rinke, C.; Hallam, S.J.; Woyke, T.; Elshahed, M.S. In Silico Analysis of the Metabolic Potential and Niche Specialization of Candidate Phylum “Latescibacteria” (WS3). PLoS ONE 2015, 10, e0127499. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; McIlroy, S.J.; Kirkegaard, R.H.; Karst, S.M.; Fernando, W.E.Y.; Aslan, H.; Meyer, R.L.; Albertsen, M.; Nielsen, P.H.; Dueholm, M.S. Novel prosthecate bacteria from the candidate phylum Acetothermia. ISME J. 2018, 12, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, J.F. The Family Ectothiorhodospiraceae. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 199–222. [Google Scholar]
- Spring, S.; Scheuner, C.; Goker, M.; Klenk, H.P. A taxonomic framework for emerging groups of ecologically important marine gammaproteobacteria based on the reconstruction of evolutionary relationships using genome-scale data. Front. Microbiol. 2015, 6, 281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, Y.-L.; Ouyang, X.; Li, J.-P.; Liao, J.-J.; You, A.; Yue, X.; Xie, G.-J.; Liang, J.-L.; Li, J.-T. Genome-Centered Metagenomics Analysis Reveals the Microbial Interactions of a Syntrophic Consortium during Methane Generation in a Decentralized Wastewater Treatment System. Appl. Sci. 2020, 10, 135. [Google Scholar] [CrossRef]
- Brümmer, I.-H.; Felske, A.-D.; Wagner-Döbler, I. Diversity and Seasonal Changes of Uncultured Planctomycetales in River Biofilms. Appl. Environ. Microbiol. 2004, 70, 5094–5101. [Google Scholar] [CrossRef]
- Iino, T.; Sakamoto, M.; Ohkuma, M. Prolixibacter denitrificans sp. nov., an iron-corroding, facultatively aerobic, nitrate-reducing bacterium isolated from crude oil, and emended descriptions of the genus Prolixibacter and Prolixibacter bellariivorans. Int. J. Syst. Evol. Microbiol. 2015, 65, 2865–2869. [Google Scholar] [CrossRef]
- Matturro, B.; Pierro, L.; Frascadore, E.; Petrangeli, P.-M.; Rossetti, S. Microbial Community Changes in a Chlorinated Solvents Polluted Aquifer Over the Field Scale Treatment With Poly-3-Hydroxybutyrate as Amendment. Front. Microbiol. 2018, 9, 1664. [Google Scholar] [CrossRef]
- Speirs, L.-B.-M.; Rice, D.-T.-F.; Petrovski, S.; Seviour, R.-J. The Phylogeny, Biodiversity, and Ecology of the Chloroflexi in Activated Sludge. Front. Microbiol. 2019, 10, 2015. [Google Scholar] [CrossRef]
- Shuman, S.; Glickman, M.-S. Bacterial DNA repair by non-homologous end joining. Nat. Rev. Microbiol. 2007, 5, 852–861. [Google Scholar] [CrossRef]
- Pitcher, R.-S.; Brissett, N.-C.; Doherty, A.-J. Nonhomologous End-Joining in Bacteria: A Microbial Perspective. Annu. Rev. Microbiol. 2007, 61, 259–282. [Google Scholar] [CrossRef]
- Sharda, M.; Badrinarayanan, A.; Seshasayee, A.-S.-N. Evolutionary and Comparative analysis of bacterial Non-Homologous End Joining Repair. Genome Biol. Evol. 2020, 12, 2450–2466. [Google Scholar] [CrossRef]
- Srinivasan, K.; Buys, E.-M. Insights into the role of bacteria in vitamin A biosynthesis: Future research opportunities. Crit. Rev. Food Sci. Nutr. 2019, 59, 3211–3226. [Google Scholar] [CrossRef]
- Choi, B.H.; Hwang, H.J.; Lee, J.E.; Oh, S.H.; Hwang, J.S.; Lee, B.Y.; Lee, P.C. Microbial Production of Retinyl Palmitate and Its Application as a Cosmeceutical. Antioxidants 2020, 9, 1130. [Google Scholar] [CrossRef]
- Oren, A. Salinibacter: An extremely halophilic bacterium with archaeal properties. FEMS Microbiol. Lett. 2013, 342, 1–9. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.-E.; Ikeda, H. Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef]
- Agger, S.A.; Lopez-Gallego, F.; Hoye, T.-R.; Schmidt-Dannert, C. Identification of Sesquiterpene Synthases from Nostoc punctiforme PCC 73102 and Nostoc sp. Strain PCC 7120. J. Bacteriol. 2008, 190, 6084–6096. [Google Scholar] [CrossRef]
- Jia, Q.; Chen, X.; Köllner, T.-G.; Rinkel, J.; Fu, J.; Labbé, J.; Xiong, W.; Dickschat, J.-S.; Gershenzon, J.; Chen, F. Terpene Synthase Genes Originated from Bacteria through Horizontal Gene Transfer Contribute to Terpenoid Diversity in Fungi. Sci. Rep. 2019, 9, 9223. [Google Scholar] [CrossRef]
- Bittman, R. Glycerolipids: Chemistry. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 25–73. [Google Scholar]
- Zhang, Y.-M.; Rock, C.-O. Thematic Review Series: Glycerolipids. Acyltransferases in bacterial glycerophospholipid synthesis. J. Lipid Res. 2008, 49, 1867–1874. [Google Scholar] [CrossRef]
- Murínová, S.; Dercová, K. Response Mechanisms of Bacterial Degraders to Environmental Contaminants on the Level of Cell Walls and Cytoplasmic Membrane. Int. J. Microbiol. 2014, 2014, 873081. [Google Scholar] [CrossRef]
- Bermingham, A.; Derrick, J.-P. The folic acid biosynthesis pathway in bacteria: Evaluation of potential for antibacterial drug discovery. BioEssays 2002, 24, 637–648. [Google Scholar] [CrossRef]
- Dick, T.; Manjunatha, U.; Kappes, B.; Gengenbacher, M. Vitamin B6 biosynthesis is essential for survival and virulence of Mycobacterium tuberculosis. Mol. Microbiol. 2010, 78, 980–988. [Google Scholar] [CrossRef]
- Cheung, M.K.; Wong, C.K.; Chu, K.H.; Kwan, H.S. Community Structure, Dynamics and Interactions of Bacteria, Archaea and Fungi in Subtropical Coastal Wetland Sediments. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Caffrey, J.; Bano, N.; Kalanetra, K.; Hollibaugh, J.T. Ammonia oxidation and ammonia-oxidizing bacteria and archaea from estuaries with differing histories of hypoxia. ISME J. 2007, 1, 660–662. [Google Scholar] [CrossRef]
- Auyeung, D.S.N.; Martiny, J.B.H.; Dukes, J.S. Nitrification kinetics and ammonia-oxidizing community respond to warming and altered precipitation. Ecosphere 2015, 6, 83. [Google Scholar] [CrossRef]
- Könneke, M.; Bernhard, A.E.; José, R.; Walker, C.B.; Waterbury, J.B.; Stahl, D.A. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 2005, 437, 543–546. [Google Scholar] [CrossRef]
- Conrad, R. The global methane cycle: Recent advances in understanding the microbial processes involved. Environ. Microbiol. Rep. 2009, 1, 285–292. [Google Scholar] [CrossRef]
- Bridgham, S.D.; Cadillo-Quiroz, H.; Keller, J.K.; Zhuang, Q. Methane emissions from wetlands: Biogeochemical, microbial, and modeling perspectives from local to global scales. Glob. Chang. Biol. 2013, 19, 1325–1346. [Google Scholar] [CrossRef] [PubMed]
- Rana, K.; Rana, N.; Singh, B. Applications of sulfur oxidizing bacteria. In Physiological and Biotechnological Aspects of Extremophiles; Salwan, R., Sharma, V., Eds.; Academic Press: London, UK, 2020; pp. 131–136. [Google Scholar]
- Koebsch, F.; Winkel, M.; Liebner, S.; Liu, B.; Westphal, J.; Schmiedinger, I.; Spitzy, A.; Gehre, M.; Jurasinski, G.; Köhler, S.; et al. Sulfate deprivation triggers high methane production in a disturbed and rewetted coastal peatland. Biogeosciences 2019, 16, 1937–1953. [Google Scholar] [CrossRef]
- Valderrama, J.A.; Durante-Rodríguez, G.; Blázquez, B.; García, J.L.; Carmona, M.; Díaz, E. Bacterial degradation of benzoate: Cross-regulation between aerobic and anaerobic pathways. J. Biol. Chem. 2012, 287, 10494–10508. [Google Scholar] [CrossRef]
- Gibson, J.; Harwood, C.S. Metabolic Diversity in Aromatic Compound Utilization by Anaerobic Microbes. Annu. Rev. Microbiol. 2002, 56, 345–369. [Google Scholar] [CrossRef] [PubMed]
- Harwood, C.S.; Burchhardt, G.; Herrmann, H.; Fuchs, G. Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol. Rev. 1998, 22, 439–458. [Google Scholar] [CrossRef]
- Zhu, J.; Tsona, N.T.; Du, L. Kinetics of atmospheric reactions of 4-chloro-1-butene. Environ. Sci. Pollut. Res. 2018, 25, 24241–24252. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-García, A.; Mestanza, O.; Isaza, J.P.; Figueroa-Galvis, I.; Vanegas, J. Influence of salinity on the degradation of xenobiotic compounds in rhizospheric mangrove soil. Environ. Pollut. 2019, 249, 750–757. [Google Scholar] [CrossRef]
- Ghosh, A.; Dey, N.; Bera, A.; Tiwari, A.; Sathyaniranjan, K.B.; Chakrabarti, K.; Chattopadhyay, D. Culture independent molecular analysis of bacterial communities in the mangrove sediment of Sundarban, India. Saline Syst. 2010, 6, 1. [Google Scholar] [CrossRef]
- Andreote, F.-D.; Jiménez, D.-J.; Chaves, D.; Dias, A.-C.; Luvizotto, D.-M.; Dini-Andreote, F.; Fasanella, C.-C.; Lopez, M.-V.; Baena, S.; Taketani, R.G.; et al. The Microbiome of Brazilian Mangrove Sediments as Revealed by Metagenomics. PLoS ONE 2012, 7, e38600. [Google Scholar] [CrossRef]
- Somboonna, N.; Assawamakin, A.; Wilantho, A.; Tangphatsornruang, S.; Tongsima, S. Metagenomic profiles of free-living archaea, bacteria and small eukaryotes in coastal areas of Sichang island, Thailand. BMC Genom. 2012, 13, S29. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Majumder, N.S.; Basak, P.; Mukherji, S.; Roy, D.; Nag, S.; Haldar, A.; Chattopadhyay, D.; Mitra, S.; Bhattacharyya, M.; et al. Diversity and Distribution of Archaea in the Mangrove Sediment of Sundarbans. Archaea 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Wang, K.; Yan, H.; Peng, X.; Hu, H.; Zhang, H.; Hou, D.; Chen, W.; Qian, P.; Liu, J.; Cai, J.; et al. Community assembly of bacteria and archaea in coastal waters governed by contrasting mechanisms: A seasonal perspective. Mol. Ecol. 2020, 29, 3762. [Google Scholar] [CrossRef]
- Ul-Hasan, S.; Bowers, R.M.; Figueroa-Montiel, A.; Licea-Navarro, A.F.; Beman, J.M.; Woyke, T.; Nobile, C.J. Community ecology across bacteria, archaea and microbial eukaryotes in the sediment and seawater of coastal Puerto Nuevo, Baja California. PLoS ONE 2019, 14, e0212355. [Google Scholar] [CrossRef]
- Liao, S.; Wang, Y.; Liu, H.; Fan, G.; Sahu, S.K.; Jin, T.; Chen, J.; Zhang, P.; Gram, L.; Strube, M.L.; et al. Deciphering the Microbial Taxonomy and Functionality of Two Diverse Mangrove Ecosystems and Their Potential Abilities To Produce Bioactive Compounds. mSystems 2020, 5, e00851-19. [Google Scholar] [CrossRef]
- Imchen, M.; Kumavath, R. Shotgun metagenomics reveals a heterogeneous prokaryotic community and a wide array of antibiotic resistance genes in mangrove sediment. FEMS Microbiol. Ecol. 2020, 96, 173. [Google Scholar] [CrossRef] [PubMed]
- Denich, T.; Beaudette, L.; Lee, H.; Trevors, J. Effect of selected environmental and physico-chemical factors on bacterial cytoplasmic membranes. J. Microbiol. Methods 2003, 52, 149–182. [Google Scholar] [CrossRef]
- Jang, H.J.; Yoon, S.H.; Ryu, H.K.; Kim, J.H.; Wang, C.H.; Kim, J.Y.; Oh, D.K.; Kim, S.W. Retinoid production using metabolically engineered Escherichia coli with a two-phase culture system. Microb. Cell Factories 2011, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Zasada, M.; Budzisz, E. Retinoids: Active molecules influencing skin structure formation in cosmetic and dermatological treatments. Postepy Dermatol. Allergol. 2019, 36, 392–397. [Google Scholar] [CrossRef]
- Hong, S.H.; Kim, K.R.; Oh, D.K. Biochemical properties of retinoid-converting enzymes and biotechnological production of retinoids. Appl. Microbiol. Biotechnol. 2015, 99, 7813–7826. [Google Scholar] [CrossRef]
- Reddy, G.K.; Leferink, N.G.H.; Umemura, M.; Ahmed, S.T.; Breitling, R.; Scrutton, N.S.; Takano, E. Exploring novel bacterial terpene synthases. PLoS ONE 2020, 15, e0232220. [Google Scholar] [CrossRef]
- Takamatsu, S.; Lin, X.; Nara, A.; Komatsu, M.; Cane, D.E.; Ikeda, H. Characterization of a silent sesquiterpenoid biosynthetic pathway in Streptomyces avermitilis controlling epi-isozizaene albaflavenone biosynthesis and isolation of a new oxidized epi-isozizaene metabolite. Microb. Biotechnol. 2011, 4, 184–191. [Google Scholar] [CrossRef]
- Martínez-Núñez, M.A.; Rodríguez-Escamilla, Z. Mining the Yucatan Coastal Microbiome for the Identification of Non-Ribosomal Peptides Synthetase (NRPS) Genes. Toxins 2020, 12, 349. [Google Scholar] [CrossRef]
- Chakraborty, R.; Coates, J.D. Anaerobic degradation of monoaromatic hydrocarbons. Appl. Microbiol. Biotechnol. 2004, 64, 437–446. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarrete-Euan, H.; Rodríguez-Escamilla, Z.; Pérez-Rueda, E.; Escalante-Herrera, K.; Martínez-Núñez, M.A. Comparing Sediment Microbiomes in Contaminated and Pristine Wetlands along the Coast of Yucatan. Microorganisms 2021, 9, 877. https://doi.org/10.3390/microorganisms9040877
Navarrete-Euan H, Rodríguez-Escamilla Z, Pérez-Rueda E, Escalante-Herrera K, Martínez-Núñez MA. Comparing Sediment Microbiomes in Contaminated and Pristine Wetlands along the Coast of Yucatan. Microorganisms. 2021; 9(4):877. https://doi.org/10.3390/microorganisms9040877
Chicago/Turabian StyleNavarrete-Euan, Herón, Zuemy Rodríguez-Escamilla, Ernesto Pérez-Rueda, Karla Escalante-Herrera, and Mario Alberto Martínez-Núñez. 2021. "Comparing Sediment Microbiomes in Contaminated and Pristine Wetlands along the Coast of Yucatan" Microorganisms 9, no. 4: 877. https://doi.org/10.3390/microorganisms9040877
APA StyleNavarrete-Euan, H., Rodríguez-Escamilla, Z., Pérez-Rueda, E., Escalante-Herrera, K., & Martínez-Núñez, M. A. (2021). Comparing Sediment Microbiomes in Contaminated and Pristine Wetlands along the Coast of Yucatan. Microorganisms, 9(4), 877. https://doi.org/10.3390/microorganisms9040877