
Molecular Epidemiology of Xanthomonas euvesicatoria Strains from the Balkan Peninsula Revealed by a New Multiple-Locus Variable-Number Tandem-Repeat Analysis Scheme


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Prediction of VNTR Loci and Primer Design
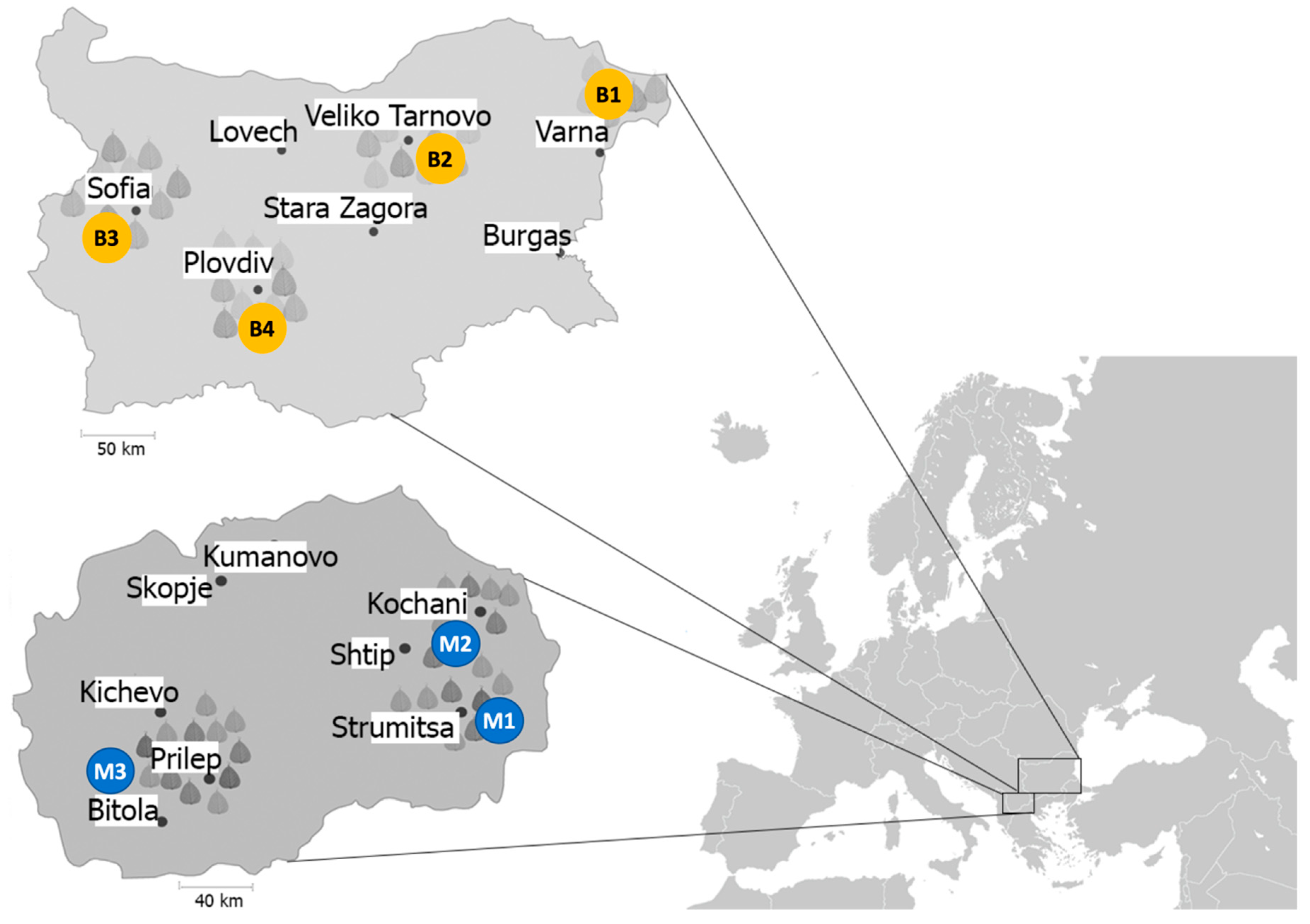
2.2. Bacterial Strains Sampling and Isolation
2.3. Molecular Biological Techniques
2.4. Multilocus Sequence Analysis
2.5. VNTR Analysis and Statistics
3. Results
3.1. Multilocus Sequence Analysis of Bulgarian and North Macedonian X. euvesicatoria Strains
3.2. Identification of Polymorphic VNTR Loci and Development of a 16-Loci MLVA Scheme (MLVA-16)
3.3. Application of the MLVA-16 Scheme on a Collection of Pepper-Pathogenic X. euvesicatoria from Bulgaria and North Macedonia
3.4. Impact of Homoplasy on VNTR Typing
3.5. Single-Nucleotide Polymorphisms in the Flanking Sequences
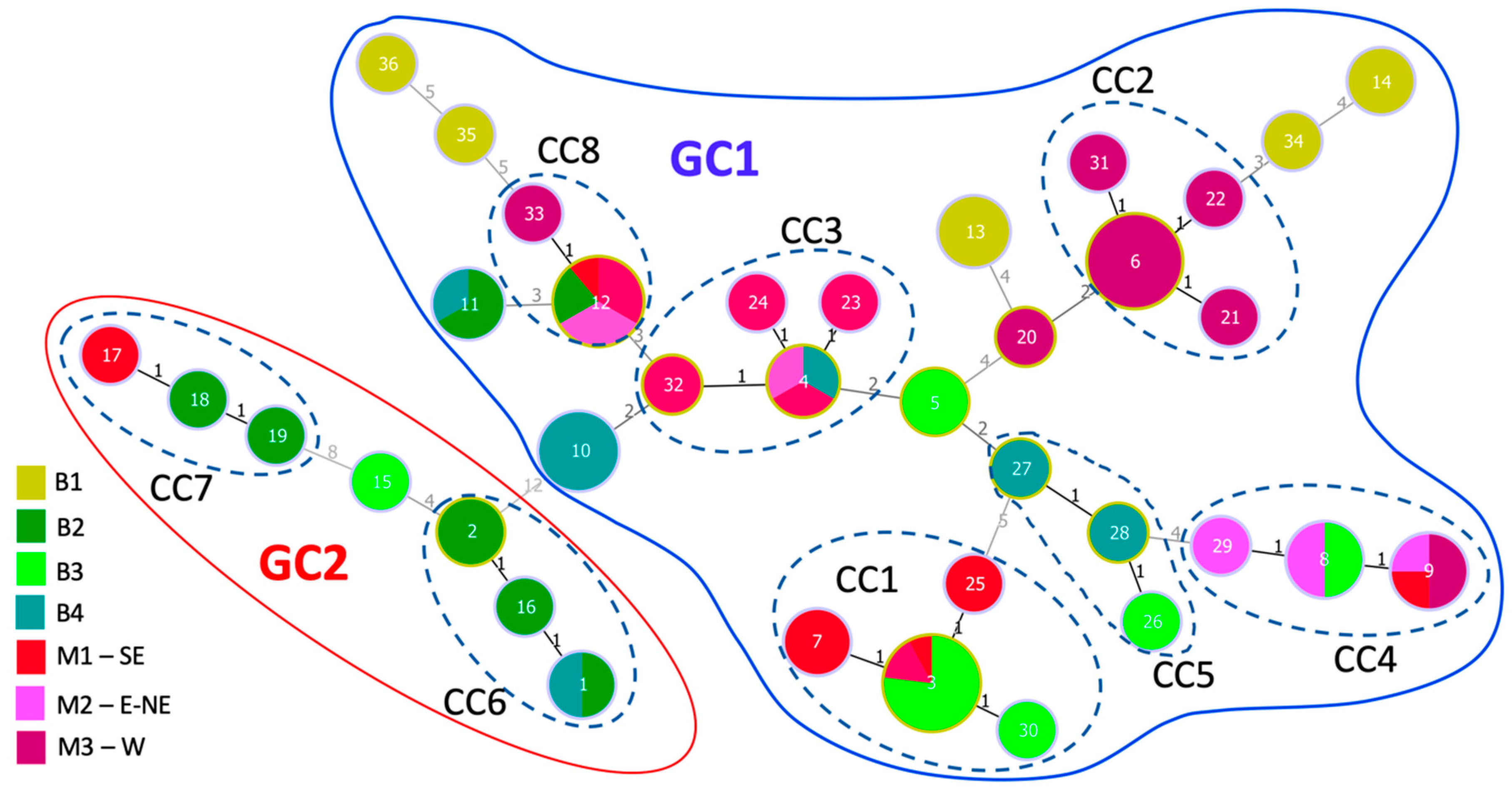
3.6. Population Structure of X. euvesicatoria
3.7. Strains from Bulgaria and North Macedonia Are Genetically Closely Related
3.8. Transferability of VNTR Markers to Other Pathovars of X. euvesicatoria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, J.B.; Stall, R.E.; Bouzar, H. Diversity among xanthomonads pathogenic on pepper and tomato. Annu. Rev. Phytopathol. 1998, 36, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Stall, R.E.; Jones, J.B.; Minsavage, G.V. Durability of resistance in tomato and pepper to xanthomonads causing bacterial spot. Annu. Rev. Phytopathol. 2009, 47, 265–284. [Google Scholar] [CrossRef]
- European and Mediterranean Plant Protection Organization. Xanthomonas spp. (Xanthomonas euvesicatoria, Xanthomonas gardneri, Xanthomonas perforans, Xanthomonas vesicatoria) causing bacterial spot of tomato and sweet pepper. EPPO Bull. 2013, 43, 7–20. [Google Scholar] [CrossRef]
- Potnis, N.; Timilsina, S.; Strayer, A.; Shantharaj, D.; Barak, J.D.; Paret, M.L.; Vallad, G.E.; Jones, J.B. Bacterial spot of tomato and pepper: Diverse Xanthomonas species with a wide variety of virulence factors posing a worldwide challenge. Mol. Plant Pathol. 2015, 16, 907–920. [Google Scholar] [CrossRef]
- Ritchie, D.F. Bacterial spot of pepper and tomato. In The Plant Health Instructor; American Phytopathology Society: St Paul, MN, USA, 2000. [Google Scholar] [CrossRef]
- Stall, R.E.; Beaulieu, C.; Egel, D.; Hodge, N.C.; Leite, R.P.; Minsavage, G.V.; Bouzar, H.; Jones, J.B.; Alvarez, A.M.; Benedict, A.A. Two genetically diverse groups of strains are included in Xanthomonas campestris pv. vesicatoria. Int. J. Syst. Bacteriol. 1994, 44, 47–53. [Google Scholar] [CrossRef]
- Dowson, W. On the systematic position and generic names of the Gram-negative bacterial plant pathogens. Zentralbl. Bakteriol. Parasitenk. Infekt. Hyg. Abt. II 1939, 100, 177–193. [Google Scholar]
- Vauterin, L.; Hoste, B.; Kersters, K.; Swings, J. Reclassification of Xanthomonas. Int. J. Syst. Bacteriol. 1995, 45, 472–489. [Google Scholar] [CrossRef]
- Šutic, D. Tomato bacteriosis. Rev. Appl. Mycol. 1957, 36, 734–735. [Google Scholar]
- Jones, J.B.; Bouzar, H.; Stall, R.E.; Almira, E.C.; Roberts, P.D.; Bowen, B.W.; Sudberry, J.; Strickler, P.M.; Chun, J. Systematic analysis of xanthomonads (Xanthomonas spp.) associated with pepper and tomato lesions. Int. J. Syst. Evol. Microbiol. 2000, 50, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Dye, D. Cultural and biochemical reaction of additional Xanthomonas species. N. Z. J. Sci. 1966, 9, 913–919. [Google Scholar]
- Jones, J.B.; Lacy, G.H.; Bouzar, H.; Stall, R.E.; Schaad, N.W. Reclassification of the xanthomonads associated with bacterial spot disease of tomato and pepper. Syst. Appl. Microbiol. 2004, 27, 755–762. [Google Scholar] [CrossRef]
- Barak, J.D.; Vancheva, T.; Lefeuvre, P.; Jones, J.B.; Timilsina, S.; Minsavage, G.V.; Vallad, G.E.; Koebnik, R. Whole-genome sequences of Xanthomonas euvesicatoria strains clarify taxonomy and reveal a stepwise erosion of type 3 effectors. Front. Plant Sci. 2016, 7, 1805. [Google Scholar] [CrossRef]
- Constantin, E.C.; Cleenwerck, I.; Maes, M.; Baeyen, S.; Van Malderghem, C.; De Vos, P.; Cottyn, B. Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016, 65, 792–806. [Google Scholar] [CrossRef]
- Oren, A.; Garrity, G.M. Notification of changes in taxonomic opinion previously published outside the IJSEM. Int. J. Syst. Evol. Microbiol. 2017, 67, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Timilsina, S.; Kara, S.; Jacques, M.-A.; Potnis, N.; Minsavage, G.V.; Vallad, G.E.; Jones, J.B.; Fischer-Le Saux, M. Reclassification of Xanthomonas gardneri (ex Šutič 1957) Jones et al., 2006 as a later heterotypic synonym of Xanthomonas cynarae Trébaol et al., 2000 and description of X. cynarae pv. cynarae and X. cynarae pv. gardneri based on whole genome analyses. Int. J. Syst. Evol. Microbiol. 2019, 69, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Morinière, L.; Burlet, A.; Rosenthal, E.R.; Nesme, X.; Portier, P.; Bull, C.T.; Lavire, C.; Fischer-Le Saux, M.; Bertolla, F. Clarifying the taxonomy of the causal agent of bacterial leaf spot of lettuce through a polyphasic approach reveals that Xanthomonas cynarae Trébaol et al., 2000 emend. Timilsina et al., 2019 is a later heterotypic synonym of Xanthomonas hortorum Vauterin et al. 1995. Syst. Appl. Microbiol. 2020, 43, 126087. [Google Scholar] [CrossRef]
- Parkinson, N.; Aritua, V.; Heeney, J.; Cowie, C.; Bew, J.; Stead, D. Phylogenetic analysis of Xanthomonas species by comparison of partial gyrase B gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2881–2887. [Google Scholar] [CrossRef]
- Parkinson, N.; Cowie, C.; Heeney, J.; Stead, D. Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int. J. Syst. Evol. Microbiol. 2009, 59, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Park, D.-C.; Shearman, H.; Fargier, E. A multilocus sequence analysis of the genus Xanthomonas. Syst. Appl. Microbiol. 2008, 31, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Hamza, A.A.; Robene-Soustrade, I.; Jouen, E.; Lefeuvre, P.; Chiroleu, F.; Fischer-Le Saux, M.; Gagnevin, L.; Pruvost, O. MultiLocus Sequence Analysis- and Amplified Fragment Length Polymorphism-based characterization of xanthomonads associated with bacterial spot of tomato and pepper and their relatedness to Xanthomonas species. Syst. Appl. Microbiol. 2012, 35, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.R.; Potnis, N.; Timilsina, S.; Wilson, M.; Patané, J.; Martins, J.J.; Minsavage, G.V.; Dahlbeck, D.; Akhunova, A.; Almeida, N.; et al. Phylogenomics of Xanthomonas field strains infecting pepper and tomato reveals diversity in effector repertoires and identifies determinants of host specificity. Front. Microbiol. 2015, 6, 535. [Google Scholar] [CrossRef] [PubMed]
- Bouzar, H.; Jones, J.; Somodi, G.; Stall, R.; Daouzli, N.; Lambe, R.; Gastelum, R.F.; Correa, R.T. Diversity of Xanthomonas campestris pv. vesicatoria in tomato and pepper fields of Mexico. Can. J. Plant Pathol. 1996, 18, 75–77. [Google Scholar] [CrossRef]
- Bouzar, H.; Jones, J.B.; Stall, R.E.; Louws, F.J.; Schneider, M.; Rademaker, J.L.W.; De Bruijn, F.J.; Jackson, L.E. Multiphasic analysis of xanthomonads causing bacterial spot disease on tomato and pepper in the Caribbean and Central America: Evidence for common lineages within and between countries. Phytopathology 1999, 89, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Quezado-Duval, A.M.; Leite, R.P., Jr.; Truffi, D.; Camargo, L.E.A. Outbreaks of bacterial spot caused by Xanthomonas gardneri on processing tomato in Central-West Brazil. Plant Dis. 2004, 88, 157–161. [Google Scholar] [CrossRef]
- Cuppels, D.A.; Louws, F.J.; Ainsworth, T. Development and evaluation of PCR-based diagnostic assays for the bacterial speck and bacterial spot pathogens of tomato. Plant Dis. 2006, 90, 451–458. [Google Scholar] [CrossRef]
- Hamza, A.A.; Robène-Soustrade, I.; Jouen, E.; Gagnevin, L.; Lefeuvre, P.; Chiroleu, F.; Pruvost, O. Genetic and pathological diversity among Xanthomonas strains responsible for bacterial spot on tomato and pepper in the Southwest Indian Ocean region. Plant Dis. 2010, 94, 993–999. [Google Scholar] [CrossRef]
- Ma, X.; Lewis Ivey, M.L.; Miller, S.A. First report of Xanthomonas gardneri causing bacterial spot of tomato in Ohio and Mich-igan. Plant Dis. 2011, 95, 1584. [Google Scholar] [CrossRef]
- Kebede, M.; Timilsina, S.; Ayalew, A.; Admassu, B.; Potnis, N.; Minsavage, G.V.; Goss, E.M.; Hong, J.C.; Strayer, A.; Paret, M.; et al. Molecular characterization of Xanthomonas strains responsible for bacterial spot of tomato in Ethiopia. Eur. J. Plant Pathol. 2014, 140, 677–688. [Google Scholar] [CrossRef]
- Araújo, E.; Costa, J.; Ferreira, M.; Quezado-Duval, A. Widespread distribution of Xanthomonas perforans and limited presence of X. gardneri in Brazil. Plant Pathol. 2016, 66, 159–168. [Google Scholar] [CrossRef]
- Osdaghi, E.; Taghavi, S.M.; Hamzehzarghani, H.; Fazliarab, A.; Lamichhane, J.R. Monitoring the occurrence of tomato bacterial spot and range of the causal agent Xanthomonas perforans in Iran. Plant Pathol. 2017, 66, 990–1002. [Google Scholar] [CrossRef]
- Osdaghi, E.; Taghavi, S.M.; Koebnik, R.; Jibrin, M.O.; Lamichhane, J.R. Multilocus sequence analysis reveals a novel phylogroup of Xanthomonas euvesicatoria pv. perforans causing bacterial spot of tomato in Iran. Plant Pathol. 2018, 67, 1601–1611. [Google Scholar] [CrossRef]
- Jones, J.B.; Pohronezny, K.L.; Stall, R.E.; Jones, J.P. Survival of Xanthomonas campestris pv. vesicatoria in Florida on tomato crop residue, weeds, seeds, and volunteer tomato plants. Phytopathology 1986, 76, 430–434. [Google Scholar] [CrossRef]
- Jones, J.B.; Stall, R.E.; Scott, J.W.; Somodi, G.C.; Bouzar, H.; Hodge, N.C. A third tomato race of Xanthomonas campestris pv. vesicatoria. Plant Dis. 1995, 79, 395–398. [Google Scholar] [CrossRef]
- Newberry, E.A.; Bhandari, R.; Minsavage, G.V.; Timilsina, S.; Jibrin, M.O.; Kemble, J.; Sikora, E.J.; Jones, J.B.; Potnis, N. Independent evolution with the gene flux originating from multiple Xanthomonas species explains genomic heterogeneity in Xanthomonas perforans. Appl. Environ. Microbiol. 2019, 85, 00885-19. [Google Scholar] [CrossRef]
- Timilsina, S.; Pereira-Martin, J.A.; Minsavage, G.V.; Iruegas-Bocardo, F.; Abrahamian, P.; Potnis, N.; Kolaczkowski, B.; Vallad, G.E.; Goss, E.M.; Jones, J.B. Multiple recombination events drive the current genetic structure of Xanthomonas perforans in Florida. Front. Microbiol. 2019, 10, 448. [Google Scholar] [CrossRef]
- EFSA Panel on Plant Health. Scientific opinion on the pest categorisation of Xanthomonas campestris pv. vesicatoria (Doidge) Dye. EFSA J. 2014, 12, 3720. [Google Scholar] [CrossRef]
- Kizheva, Y.; Vancheva, T.; Hristova, P.; Stoyanova, M.; Stojanovska, M.; Moncheva, P.; Bogatzevska, N. Identification of Xanthomonas strains from tomato and pepper and their sensitivity to antibiotics and copper. Bulg. J. Agric. Sci. 2013, 19, 80–82. [Google Scholar]
- EPPO A2 List of Pests Recommended for Regulation as Quarantine Pests. Available online: https://www.eppo.int/ACTIVITIES/plant_quarantine/A2_list (accessed on 27 November 2020).
- Buonaurio, R.; Stravato, V.M.; Scortichini, M. Characterization of Xanthomonas campestris pv. vesicatoria from Capsicum annuum L. in Italy. Plant Dis. 1994, 78, 296–299. [Google Scholar] [CrossRef]
- Zaccardelli, M.; Campanile, F.; Villecco, D.; Parisi, M. Infections of bacterial spot on processing tomato in southern Italy. ISHS Acta Hortic. 2011, 914, 71–73. [Google Scholar] [CrossRef]
- Pernezny, K.; Kůdela, V.; Kokošková, B.; Hládká, I. Bacterial diseases of tomato in the Czech and Slovak Republics and lack of streptomycin resistance among copper-tolerant bacterial strains. Crop. Prot. 1995, 14, 267–270. [Google Scholar] [CrossRef]
- Beran, P.; Mraz, I.; Kokoskova, B.; Bohata, A. Monitoring the occurrence of bacterial spot of tomato and pepper in the Czech Republic and development of new PCR primers for detection of Xanthomonas vesicatoria. Eur. J. Plant Pathol. 2015, 141, 617–621. [Google Scholar] [CrossRef]
- Obradovic, A.; Mavridis, A.; Rudolph, K.; Arsenijević, M. Bacterial spot of capsicum and tomato in Yugoslavia. EPPO Bull. 2000, 30, 333–336. [Google Scholar] [CrossRef]
- Kovachevski, I. Trudove na bulgarskoto prirodoizpitatelno druzhestvo. Sci. Works Bulg. Nat. Soc. 1936, 17, 13–24. [Google Scholar]
- Karov, S. Xanthomonas vesicatoria (Doidge) Dowson of pepper in Bulgaria. Bulg. Nat. Soc. 1965, 14, 245–250. [Google Scholar]
- Mitrev, S.; Pejcinovski, F. Characterization of Xanthomonas campestris pv. vesicatoria, causal agent of bacterial spot of pepper, cv. Kurtovska kapija. In Yearbook for Plant Protection; Society for Plant Protection of Republic of Macedonia: Skopje, Macedonia, 1999; Volume X, pp. 151–163. [Google Scholar]
- Mitrev, S. Phytopathogenic Bacteria of Pepper in Macedonia; PSI Institute of Southern Crops Strumica: Strumica, Macedonia, 2001. [Google Scholar]
- Bogatzevska, N.; Stoimenova, E.; Mitrev, E. Bacterial and virus diseases spread in Bulgaria and Macedonia on field and greenhouse pepper. Plant Prot. 2007, 18, 17–21. [Google Scholar]
- Kizheva, Y.; Vancheva, T.; Hristova, P.; Stoyanova, M.; Bogatzevska, N.; Moncheva, P. Diversity of Xanthomonas spp. causal agents of bacterial spot on pepper and tomato plants in Bulgaria. Biotechnol. Biotechnol. Equip. 2011, 25, 98–104. [Google Scholar] [CrossRef]
- Kizheva, Y.; Vancheva-Ebben, T.; Hristova, P.; Bogatzevska, N.; Moncheva, P. First report of Xanthomonas euvesicatoria on tomato in Bulgaria. C. R. Acad. Bulg. Sci. 2020, 73, 140–146. [Google Scholar] [CrossRef]
- Vancheva, T.; Stoyanova, M.; Tatyozova, M.; Bogatzevska, N.; Moncheva, P. Sub-species diversity of Xanthomonas euvesicatoria Bulgarian and Macedonian strains from pepper. Biotechnol. Biotechnol. Equip. 2014, 28, 592–601. [Google Scholar] [CrossRef]
- Vancheva, T.; Stoyanova, M.; Tasheva-Terzieva, E.; Bogatzevska, N.; Moncheva, P. Molecular methods for diversity assessment among xanthomonads of Bulgarian and Macedonian pepper. Braz. J. Microbiol. 2018, 49, 246–259. [Google Scholar] [CrossRef]
- Timilsina, S.; Jibrin, M.O.; Potnis, N.; Minsavage, G.V.; Kebede, M.; Schwartz, A.; Bart, R.; Staskawicz, B.; Boyer, C.; Vallad, G.E.; et al. Multilocus sequence analysis of xanthomonads causing bacterial spot of tomato and pepper plants reveals strains generated by recombination among species and recent global spread of Xanthomonas gardneri. Appl. Environ. Microbiol. 2015, 81, 1520–1529. [Google Scholar] [CrossRef]
- Bui Thi Ngoc, L.; Vernière, C.; Vital, K.; Guérin, F.; Gagnevin, L.; Brisse, S.; Ah-You, N.; Pruvost, O. Fourteen minisatellite markers for population studies of the citrus canker bacterium, Xanthomonas citri pv. citri. Mol. Ecol. Res. 2009, 9, 125–127. [Google Scholar] [CrossRef]
- Pruvost, O.; Vernière, C.; Vital, K.; Guérin, F.; Jouen, E.; Chiroleu, F.; Ah-You, N.; Gagnevin, L. Insertion sequence- and tandem repeat-based genotyping techniques for Xanthomonas citri pv. mangiferaeindicae. Phytopathology 2011, 101, 887–893. [Google Scholar] [CrossRef]
- Gironde, S.; Manceau, C. Housekeeping gene sequencing and multilocus variable-number tandem-repeat analysis to identify subpopulations within Pseudomonas syringae pv. maculicola and Pseudomonas syringae pv. tomato that correlate with host specificity. Appl. Environ. Microbiol. 2012, 78, 3266–3279. [Google Scholar] [CrossRef]
- Zhao, S.; Poulin, L.; Rodriguez-R, L.M.; Serna, N.F.; Liu, S.-Y.; Wonni, I.; Szurek, B.; Verdier, V.; Leach, J.E.; He, Y.-Q.; et al. Development of a variable number of tandem repeats typing scheme for the bacterial rice pathogen Xanthomonas oryzae pv. oryzicola. Phytopathology 2012, 102, 948–956. [Google Scholar] [CrossRef]
- N’Guessan, C.A.; Brisse, S.; Le Roux-Nio, A.-C.; Poussier, S.; Kone, D.; Wicker, E. Development of variable number of tandem repeats typing schemes for Ralstonia solanacearum, the agent of bacterial wilt, banana Moko disease and potato brown rot. J. Microbiol. Methods 2013, 92, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Cesbron, S.; Pothier, J.; Gironde, S.; Jacques, M.-A.; Manceau, C. Development of multilocus variable-number tandem repeat analysis (MLVA) for Xanthomonas arboricola pathovars. J. Microbiol. Methods 2014, 100, 84–90. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Arias-Rojas, N.; Poulin, L.; A Medina, C.; Tapiero, A.; Restrepo, S.; Koebnik, R.; Bernal, A.J. Population typing of the causal agent of cassava bacterial blight in the Eastern Plains of Colombia using two types of molecular markers. BMC Microbiol. 2014, 14, 161. [Google Scholar] [CrossRef]
- Poulin, L.; Grygiel, P.; Magne, M.; Gagnevin, L.; Rodríguez-R, L.M.; Serna, N.F.; Zhao, S.; El Rafii, M.; Dao, S.; Tekete, C.; et al. New multilocus variable-number tandem-repeat analysis tool for surveillance and local epidemiology of bacterial leaf blight and bacterial leaf streak of rice caused by Xanthomonas oryzae. Appl. Environ. Microbiol. 2015, 81, 688–698. [Google Scholar] [CrossRef]
- López-Soriano, P.; Boyer, K.; Cesbron, S.; Morente, M.C.; Peñalver, J.; Palacio-Bielsa, A.; Vernière, C.; López, M.M.; Pruvost, O. Multilocus variable number of tandem repeat analysis reveals multiple introductions in Spain of Xanthomonas arboricola pv. pruni, the causal agent of bacterial spot disease of stone fruits and almond. PLoS ONE 2016, 11, e0163729. [Google Scholar] [CrossRef]
- Gétaz, M.; Krijger, M.; Rezzonico, F.; Smits, T.H.M.; Van Der Wolf, J.M.; Pothier, J.F. Genome-based population structure analysis of the strawberry plant pathogen Xanthomonas fragariae reveals two distinct groups that evolved independently before its species description. Microb. Genom. 2018, 4, e000189. [Google Scholar] [CrossRef] [PubMed]
- Rache, L.; Blondin, L.; Flores, C.; Trujillo, C.; Szurek, B.; Restrepo, S.; Koebnik, R.; Bernal, A.; Vernière, C. An optimized microsatellite scheme for assessing populations of Xanthomonas phaseoli pv. manihotis. Phytopathology 2019, 109, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [CrossRef]
- Estoup, A.; Jarne, P.; Cornuet, J.-M. Homoplasy and mutation model at microsatellite loci and their consequences for population genetics analysis. Mol. Ecol. 2002, 11, 1591–1604. [Google Scholar] [CrossRef]
- Valdes, A.M.; Slatkin, M.; Freimer, N.B. Allele frequencies at microsatellite loci: The stepwise mutation model revisited. Genetics 1993, 133, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Jarne, P.; Lagoda, P.J. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 1996, 11, 424–429. [Google Scholar] [CrossRef]
- Angers, B.; Estoup, A.; Jarne, P. Microsatellite size homoplasy, SSCP, and population structure: A case study in the freshwater snail Bulinus truncatus. Mol. Biol. Evol. 2000, 17, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Thieme, F.; Koebnik, R.; Bekel, T.; Berger, C.; Boch, J.; Büttner, D.; Caldana, C.; Gaigalat, L.; Goesmann, A.; Kay, S.; et al. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium Xanthomonas campestris pv. vesicatoria revealed by the complete genome sequence. J. Bacteriol. 2005, 187, 7254–7266. [Google Scholar] [CrossRef] [PubMed]
- Vancheva, T.; Lefeuvre, P.; Bogatzevska, N.; Moncheva, P.; Koebnik, R. Draft genome sequences of two Xanthomonas euvesicatoria strains from the balkan peninsula. Genome Announc. 2015, 3, e01528-14. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, J.L.; Hoste, B.; Louws, F.J.; Kersters, K.; Swings, J.; Vauterin, L.; De Bruijn, F.J. Comparison of AFLP and rep-PCR genomic fingerprinting with DNA-DNA homology studies: Xanthomonas as a model system. Int. J. Syst. Evol. Microbiol. 2000, 50, 665–677. [Google Scholar] [CrossRef]
- Rademaker, J.L.W.; Louws, F.J.; Schultz, M.H.; Rossbach, U.; Vauterin, L.; Swings, J.; De Bruijn, F.J. A comprehensive species to strain taxonomic framework for Xanthomonas. Phytopathology 2005, 95, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Jalan, N.; Aritua, V.; Kumar, D.; Yu, F.; Jones, J.B.; Graham, J.H.; Setubal, J.C.; Wang, N. Comparative genomic analysis of Xanthomonas axonopodis pv. citrumelo F1, which causes citrus bacterial spot disease, and related strains provides insights into virulence and host specificity. J. Bacteriol. 2011, 193, 6342–6357. [Google Scholar] [CrossRef] [PubMed]
- Potnis, N.; Krasileva, K.; Chow, V.; Almeida, N.F.; Patil, P.B.; Ryan, R.P.; Sharlach, M.; Behlau, F.; Dow, J.M.; Momol, M.; et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genom. 2011, 12, 146. [Google Scholar] [CrossRef]
- Jacques, M.-A.; Bolot, S.; Charbit, E.; Darrasse, A.; Briand, M.; Arlat, M.; Gagnevin, L.; Koebnik, R.; Noël, L.D.; Portier, P.; et al. High-quality draft genome sequence of Xanthomonas alfalfae subsp. alfalfae strain CFBP 3836. Genome Announc. 2013, 1, 01035-13. [Google Scholar] [CrossRef] [PubMed]
- Gagnevin, L.; Bolot, S.; Gordon, J.L.; Pruvost, O.; Vernière, C.; Robene, I.; Arlat, M.; Noël, L.D.; Carrère, S.; Jacques, M.-A.; et al. Draft genome sequence of Xanthomonas axonopodis pv. allii strain CFBP 6369. Genome Announc. 2014, 2, e00727-14. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carriço, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinformatics 2012, 13, 87. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Kalinowski, S.T. hp-rare 1.0: A computer program for performing rarefaction on measures of allelic diversity. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Jibrin, M.O.; Timilsina, S.; Potnis, N.; Minsavage, G.V.; Shenge, K.C.; Akpa, A.D.; Alegbejo, M.D.; Beed, F.; Vallad, G.E.; Jones, J.B. First report of atypical Xanthomonas euvesicatoria strains causing bacterial spot of tomato in Nigeria. Plant Dis. 2015, 99, 415. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, U.; Dobhal, S.; Alvarez, A.M.; Arif, M. Phylogenetic analyses of xanthomonads causing bacterial leaf spot of tomato and pepper: Xanthomonas euvesicatoria revealed homologous populations despite distant geographical distribution. Microorganisms 2019, 7, 462. [Google Scholar] [CrossRef]
- Almeida, N.F.; Yan, S.; Cai, R.; Clarke, C.R.; Morris, C.E.; Schaad, N.W.; Schuenzel, E.L.; Lacy, G.H.; Sun, X.; Jones, J.B.; et al. PAMDB, a multilocus sequence typing and analysis database and website for plant-associated microbes. Phytopathology 2000, 100, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Taylor, K.W.; Mudgett, M.B. Comparative analysis of the XopD type III secretion (T3S) effector family in plant pathogenic bacteria. Mol. Plant Pathol. 2011, 12, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Louws, F.; Rademaker, J.; De Bruijn, F. The three Ds of PCR-based genomic analysis of phytobacteria: Diversity, detection, and disease diagnosis. Annu. Rev. Phytopathol. 1999, 37, 81–125. [Google Scholar] [CrossRef] [PubMed]
- Pruvost, O.; Magne, M.; Boyer, K.; LeDuc, A.; Tourterel, C.; Drevet, C.; Ravigné, V.; Gagnevin, L.; Guérin, F.; Chiroleu, F.; et al. A MLVA genotyping scheme for global surveillance of the citrus pathogen Xanthomonas citri pv. citri suggests a worldwide geographical expansion of a single genetic lineage. PLoS ONE 2014, 9, e98129. [Google Scholar] [CrossRef]
- Essakhi, S.; Cesbron, S.; Fischer-Le Saux, M.; Bonneau, S.; Jacques, M.-A.; Manceau, C. Phylogenetic and variable-number tandem-repeat analyses identify nonpathogenic Xanthomonas arboricola lineages lacking the canonical type III secretion system. Appl. Environ. Microbiol. 2015, 81, 5395–5410. [Google Scholar] [CrossRef] [PubMed]
- Leduc, A.; Traoré, Y.N.; Boyer, K.; Magne, M.; Grygiel, P.; Juhasz, C.C.; Boyer, C.; Guerin, F.; Wonni, I.; Ouedraogo, L.; et al. Bridgehead invasion of a monomorphic plant pathogenic bacterium: Xanthomonas citri pv. citri, an emerging citrus pathogen in Mali and Burkina Faso. Environ. Microbiol. 2015, 17, 4429–4442. [Google Scholar] [CrossRef]
- Ferreira, M.A.S.V.; Bonneau, S.; Briand, M.; Cesbron, S.; Portier, P.; Darrasse, A.; Gama, M.A.S.; Barbosa, M.A.G.; Mariano, R.D.L.R.; Souza, E.B.; et al. Xanthomonas citri pv. viticola affecting grapevine in Brazil: Emergence of a successful monomorphic pathogen. Front. Plant Sci. 2019, 10, 489. [Google Scholar] [CrossRef]
- Nakato, G.V.; Rojas, J.L.F.; Vernière, C.; Blondin, L.; Coutinho, T.; Mahuku, G.; Wicker, E. A new Multi Locus Variable Number of Tandem Repeat Analysis Scheme for epidemiological surveillance of Xanthomonas vasicola pv. musacearum, the plant pathogen causing bacterial wilt on banana and enset. PLoS ONE 2019, 14, e0215090. [Google Scholar] [CrossRef] [PubMed]
- Bui Thi Ngoc, L.; Vernière, C.; Jarne, P.; Brisse, S.; Guérin, F.; Boutry, S.; Gagnevin, L.; Pruvost, O.; Guérin, F. From local surveys to global surveillance: Three high-throughput genotyping methods for epidemiological monitoring of Xanthomonas citri pv. citri pathotypes. Appl. Environ. Microbiol. 2008, 75, 1173–1184. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schlötterer, C. Evolutionary dynamics of microsatellite DNA. Chromosoma 2000, 109, 365–371. [Google Scholar] [CrossRef]
- Jibrin, M.O.; Potnis, N.; Timilsina, S.; Minsavage, G.V.; Vallad, G.E.; Roberts, P.D.; Jones, J.B.; Goss, E.M. Genomic inference of recombination-mediated evolution in Xanthomonas euvesicatoria and X. perforans. Appl. Environ. Microbiol. 2018, 84, e00136-18. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Locus | Position in Strain 85-10 1 | Dominant Repeat Type | Other Repeat Types 2 | No. of Alleles 3 | Allelic Range 4 | HGDI Score 5 |
|---|---|---|---|---|---|---|
| Xe_02 | 215122..215170 | TCCCCAT | - | 4 | 4–7 # | 0.286 |
| Xe_03 | 487069..487152 | TTTGGC | TCTGGC * TTCGGC TTTGTC * | 3 | 12–14 # | 0.190 |
| Xe_04 | 624229..624277 | CGATTCC | - | 8 | 5–12# | 0.764 |
| Xe_06 | 857148..857196 | AACAGCC | - | 3 | 6–8 # | 0.317 |
| Xe_07 | 924719..924767 | CCGGGTC | CCGGGCC * | 4 | 4–7 # | 0.211 |
| Xe_09 | 1053822..1053863 | GGGATTT | GGGATTC GGGAATC | 7 | 6–18 | 0.803 |
| Xe_10 | 1222069..1222110 | AGGCGGT | AGGCGGC * | 6 | 5–12 | 0.575 |
| Xe_11 | 1504314..1504418 | CCGATTC | CCTAATC CCCAATC | 5 (6) | 11–16 | 0.455 (0.480) |
| Xe_14 | 2268785..2268850 | ACAGCG | - | 6 | 6–11 # | 0.738 |
| Xe_15 | 3198440..3198527 | GCAGACAG | GCAGGCAG GCAGAGAT * | 5 (8) | 6–10 # | 0.688 (0.779) |
| Xe_16 | 3505639..3505687 | AATGGGG | AATCGGG * | 3 | 5–9 | 0.263 |
| Xe_17 | 3514941..3514994 | TCGGCA | TCGGCG * | 5 | 9–14 | 0.437 |
| Xe_22 | 4396287..4396335 | TTGGCGG | TTGGCGC * | 3 | 5–10 | 0.190 |
| Xe_29 | 4211581..4211608 | CGATTCC | - | 2 | 4–5 # | 0.315 |
| Xe_34 | 458055..458096 | GATTCGG | GAATCGG GAATTCG * GAATCCG * | 4 (5) | 5–16 | 0.190 (0.192) |
| Xe_49 | 4410313..4410342 | TGGCCG | - | 4 | 5–8# | 0.575 |
| MLVA-16 | 36 haplotypes | 0.944 |
| VNTR Locus | No. of Repeats | ST | Strains 1 | Repeat Pattern |
|---|---|---|---|---|
| Xe_11 | 13 | 1 | CC6 (62b, 67b, 74b, 96b, 102b)/CC7 (42b, 43b, 35M)/ MT15 (13b) | (CCGATTC)7-(CCCAATC)1-(CCTAATC)1-(CCCAATC)4 |
| Xe_11 | 13 | 2 | CC3 (10b, 1M, 2M, 5M, 25M, 50M)/MT10 (44b, 45b, 47b, 49b, 51b) | (CCGATTC)6-(CCCAATC)1-(CCTAATC)1-(CCCAATC)5 |
| Xe_15 | 10 | 1 | CC3 (10b, 1M, 2M, 5M, 25M, 50M)/MT11 (61b, 69b, 70b) | (GCAGGCAG)3-(GCAGACAG)6-(GCAGAGAT)1 |
| Xe_15 | 10 | 2 | CC8 (105b, 106b, 77M, 79M, 80M, 81M, 82M, 83M, 84M, 86M)/MT35 (80b) | (GCAGGCAG)2-(GCAGACAG)7-(GCAGAGAT)1 |
| Xe_15 | 9 | 1 | CC1 (5b, 12b, 24b, 27b, 29b, 30b, 31b, 38b, 55b, 56b, 7M, 11M, 28M, 31M, 37M, 38M)/ CC5 (39b, 93b, 94b)/MT5 (11b, 54b)/MT10 (44b, 45b, 47b, 49b, 51b)/MT14 (81b, 82b)/MT34 (85b)/MT36 (86b) | (GCAGGCAG)2-(GCAGACAG)6-(GCAGAGAT)1 |
| Xe_15 | 9 | 2 | CC6 (62b, 67b, 74b, 96b, 102b) | (GCAGGCAG)1-(GCAGACAG)7-(GCAGAGAT)1 |
| Xe_15 | 8 | 1 | CC1 (28b)/MT13 (77b, 78b, 79b) | (GCAGGCAG)2-(GCAGACAG)5-(GCAGAGAT)1 |
| Xe_15 | 8 | 2 | CC2 (54M, 55M, 56M, 57M, 58M, 61M, 62M, 63M,64M, 65M, 66M, 67M, 68M, 69M, 70M)/CC4 (89b, 90b, 71M, 72M, 73M, 74M, 76M, 85M, 87M)/MT20 (59M) | (GCAGGCAG)3-(GCAGACAG)4-(GCAGAGAT)1 |
| Xe_34 | 13 | 1 | CC7 (42b, 35M) | (GATTCGG)5-(GAATCGG)2-(GATTCGG)1-(GAATCGG)4-(GAATTCG)1 |
| Xe_34 | 13 | 2 | CC6 (62b, 67b, 74b, 96b, 102b) | (GATTCGG)4-(GAATCGG)2-(GATTCGG)1-(GAATCGG)5-(GAATTCG)1 |
| Country | Polymorphic Loci | eMLG 1 | Simpson Index D | HE 2 (seq) 4 | A 3 (seq) 4 | Ap 3 (seq) 4 |
|---|---|---|---|---|---|---|
| Both countries | 22.9 | 0.944 | 0.437 | - | - | |
| Bulgaria | 16 | 21.5 | 0.932 | 0.494 (0.506) | 4.25 (4.56) | 1.25 (1.44) |
| North Macedonia | 15 5 | 18 | 0.890 | 0.329 (0.332) | 3.13 (3.25) | 0.13 (0.13) |
| Source of Variation | D.f. | Sum of Squares | Variance Components | Percentage of Variation | p-Value |
|---|---|---|---|---|---|
| - between countries | 1 | 177.11 | 0.329 | 1.05 | 0.437 |
| - between regions within countries | 5 | 706.15 | 10.05 | 32.14 | <0.001 |
| - within regions | 81 | 1691.68 | 20.88 | 66.80 | <0.001 |
| Total | 87 | 2574.94 | 31.26 |
| B2 | B3 | B4 | M1 | M2 | M3 | |
|---|---|---|---|---|---|---|
| B1 | 0.442 ** | 0.304 *** | 0.239 ** | 0.250 ** | 0.321 * | 0.501 *** |
| B2 | 0.400 ** | 0.358 ** | 0.406 ** | 0.447 * | 0.645 *** | |
| B3 | 0.012 NS | −0.031 NS | 0.070 NS | 0.313 *** | ||
| B4 | −0.040 NS | −0.014 NS | 0.200 ** | |||
| M1 | −0.001 NS | 0.237 *** | ||||
| M2 | 0.451 *** |
| Locus | euvesicatoria | perforans | alfalfae | allii | citrumelo | commiphoreae | dieffenbachiae |
|---|---|---|---|---|---|---|---|
| Xe_02 | 2–11 | 1–16 | 13 | 10 | 12 | 18 | 15 |
| Xe_03 | 8–14 | 11–12 | NA | NA | NA | NA | NA |
| Xe_04 | 3–11 | 2–3 | 3 | 9 | 3–4 | 4 | 4 |
| Xe_06 | 3–11 | 2 | 2 | 4 | 2 | 6 | 2 |
| Xe_07 | 3–8 | 4 | 2 | 4 | 2–8 | 6 | 6 |
| Xe_09 | 5–16 | 5–7 (11) | 5-6 | 12 | 12–13 | 6 | 14 |
| Xe_10 | 3–8 | (3) 5–7 | 4 | 4 | 4 | 5 | 10 |
| Xe_11 | 9–17 | 4–6 | 4 | 9 | 5–6 | 9 | 4 |
| Xe_14 | 2–13 | 5–8 | 5 | 9 | 8–9 | 6 | 9 |
| Xe_15 | 6–11 | 2 (6.5) | 2 | 2 | 4–6 | 6 | 2 |
| Xe_16 | 3–16 | (4) 10–12 | 12 | 4 | 4 | 4 | 11 |
| Xe_17 | 8–18 | 10–16 | 9 | 15 | 9–12 | 15 | 10 |
| Xe_22 | 5–21 | 3–9 | 5 | 12 | 4 | 16 | 8 |
| Xe_29 | 3–4 | 4–7 | 2–9 | 5 | 5 | 4 | 3 |
| Xe_34 | 4–15 | 9–14 | 7–12 | 8 | 8–11 | 15 | 7 |
| Xe_49 | 7–18 | (4) 6–7 | 6–7 | 4 | 9–15 | 10 | 9 |
| No. of strains | 54 | 143 | 2 | 1 | 4 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vancheva, T.; Bogatzevska, N.; Moncheva, P.; Mitrev, S.; Vernière, C.; Koebnik, R. Molecular Epidemiology of Xanthomonas euvesicatoria Strains from the Balkan Peninsula Revealed by a New Multiple-Locus Variable-Number Tandem-Repeat Analysis Scheme. Microorganisms 2021, 9, 536. https://doi.org/10.3390/microorganisms9030536
Vancheva T, Bogatzevska N, Moncheva P, Mitrev S, Vernière C, Koebnik R. Molecular Epidemiology of Xanthomonas euvesicatoria Strains from the Balkan Peninsula Revealed by a New Multiple-Locus Variable-Number Tandem-Repeat Analysis Scheme. Microorganisms. 2021; 9(3):536. https://doi.org/10.3390/microorganisms9030536
Chicago/Turabian StyleVancheva, Taca, Nevena Bogatzevska, Penka Moncheva, Sasa Mitrev, Christian Vernière, and Ralf Koebnik. 2021. "Molecular Epidemiology of Xanthomonas euvesicatoria Strains from the Balkan Peninsula Revealed by a New Multiple-Locus Variable-Number Tandem-Repeat Analysis Scheme" Microorganisms 9, no. 3: 536. https://doi.org/10.3390/microorganisms9030536
APA StyleVancheva, T., Bogatzevska, N., Moncheva, P., Mitrev, S., Vernière, C., & Koebnik, R. (2021). Molecular Epidemiology of Xanthomonas euvesicatoria Strains from the Balkan Peninsula Revealed by a New Multiple-Locus Variable-Number Tandem-Repeat Analysis Scheme. Microorganisms, 9(3), 536. https://doi.org/10.3390/microorganisms9030536