
Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Drugs
2.3. Host Cells Cultures
2.4. Culture of Parasites
2.5. T. cruzi Growth Inhibition Assay
2.6. Anti-Amastigote Specific Assay
2.7. NIH-3T3 Cells-Based Assays
2.8. Vero and HepG2 Toxicity Assays
2.9. T. cruzi In Vivo Inhibition Assay
2.10. Data Analysis
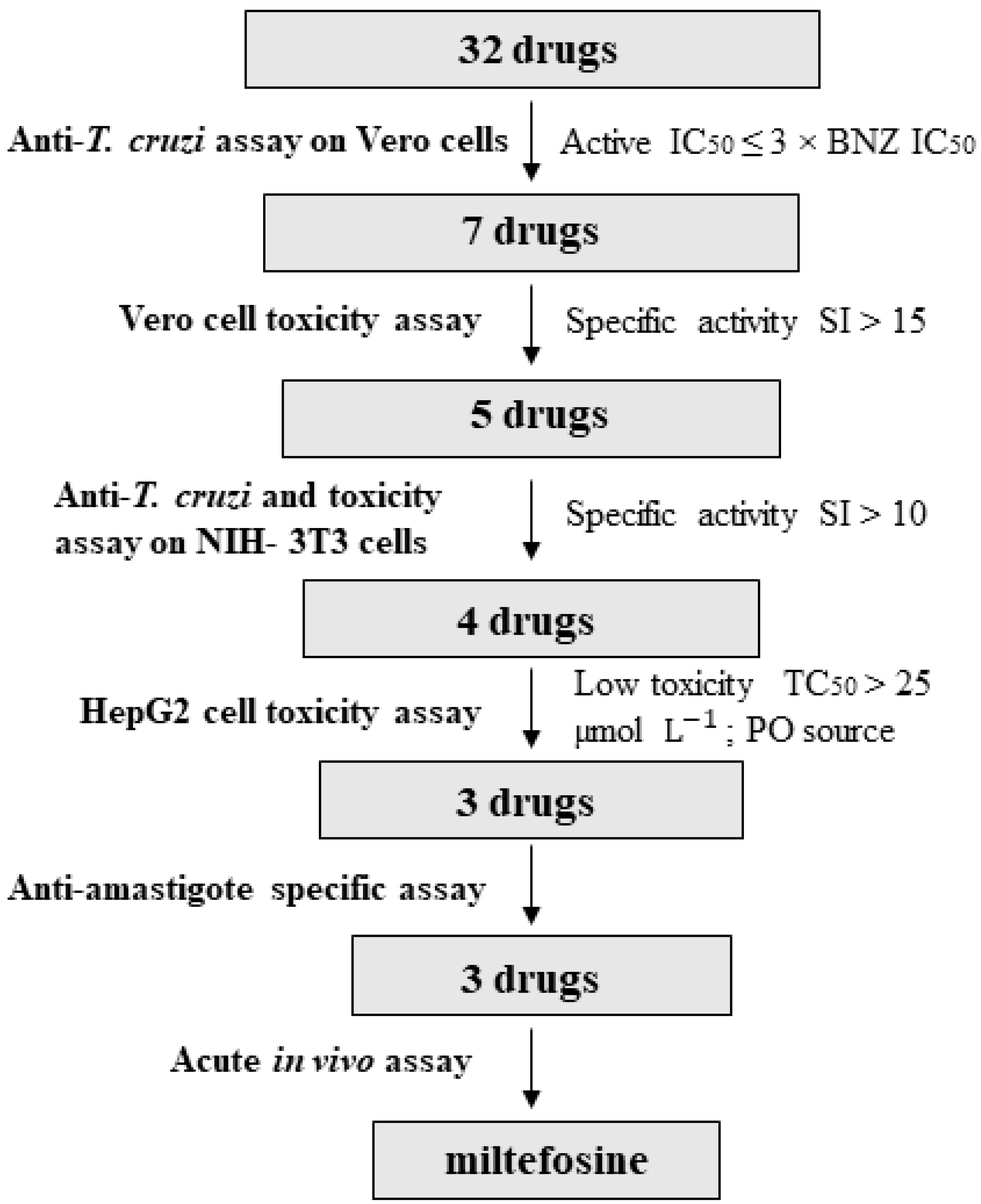
3. Results
3.1. Anti-T. cruzi Activity on Vero Cells
3.2. Identification of Drugs with Specific Activity against the Parasite
3.3. Anti-T. cruzi Growth Inhibition of Active Drugs Confirmed
3.4. HepG2 Toxicity Assay
3.5. Anti-Amastigote Specific Activity of Selected Drugs
3.6. In Vivo Anti-T. cruzi Activity of the Selected Drugs in a Mouse Model of Acute Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO: Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/en/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 26 January 2020).
- Gascon, J.; Bern, C.; Pinazo, M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Fabbro, D.L.; Streiger, M.L.; Arias, E.D.; Bizai, M.L.; Del Barco, M.; Amicone, N.A. Trypanocide treatment among adults with chronic Chagas disease living in Santa Fe City (Argentina), over a mean follow-up of 21 years: Parasitological, serological and clinical evolution. Rev. Soc. Bras. Med. Trop. 2007, 40, 1–10. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Cortés-Serra, N.; Pinazo, M.J.; Elena, M.; Abril, M.; Barreira, F.; Sosa-Estani, S.; Hotez, P.J.; Gascón, J. Strategies to enhance access to diagnosis and treatment for Chagas disease patients in Latin America. Expert Rev. Anti. Infect. Ther. 2019, 17, 145–157. [Google Scholar] [CrossRef]
- Crespillo, C.; Emmanuele, A.; Rullo, V.; López, R.; Begoña, V.; Maillo, M. Safety profile of benznidazole in the treatment of chronic Chagas disease: Experience of a referral centre and systematic literature review with meta-analysis. Drug Saf. 2018, 41, 1035–1048. [Google Scholar] [CrossRef]
- Forsyth, C.J.; Hernandez, S.; Olmedo, W.; Abuhamidah, A.; Traina, M.I.; Sanchez, D.R.; Soverow, J.; Meymandi, S.K. Safety profile of nifurtimox for treatment of Chagas disease in the United States. Clin. Infect. Dis. 2016, 63, 1056–1062. [Google Scholar] [CrossRef]
- Pinazo, M.J.; Muñoz, J.; Posada, E.; López-Chejade, P.; Gállego, M.; Ayala, E.; Del Cacho, E.; Soy, D.; Gascon, J. Tolerance of benznidazole in treatment of Chagas’ disease in adults. Antimicrob. Agents Chemother. 2010, 54, 4896–4899. [Google Scholar] [CrossRef]
- Jackson, Y.; Alirol, E.; Getaz, L.; Wolff, H.; Combescure, C.; Chappuis, F. Tolerance and safety of nifurtimox in patients with chronic Chagas disease. Clin. Infect. Dis. 2010, 51, e69–e75. [Google Scholar] [CrossRef]
- Urbina, J.A. New insights in Chagas’ disease treatment. Drugs Future 2010, 35, 409–419. [Google Scholar] [CrossRef]
- Khare, S.; Nagle, A.S.; Biggart, A.; Lai, Y.H.; Liang, F.; Davis, L.C.; Barnes, S.W.; Mathison, C.J.N.; Myburgh, E.; Gao, M.Y.; et al. Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 2016, 537, 229–233. [Google Scholar] [CrossRef]
- Peña, I.; Pilar Manzano, M.; Cantizani, J.; Kessler, A.; Alonso-Padilla, J.; Bardera, A.I.; Alvarez, E.; Colmenarejo, G.; Cotillo, I.; Roquero, I.; et al. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015, 5, 8771. [Google Scholar]
- Martínez-Peinado, N.; Cortes-Serra, N.; Losada-Galvan, I.; Alonso-Vega, C.; Urbina, J.A.; Rodríguez, A.; VandeBerg, J.L.; Pinazo, M.J.; Gascon, J.; Alonso-Padilla, J. Emerging agents for the treatment of Chagas disease: What is in the preclinical and clinical development pipeline? Expert Opin. Investig. Drugs 2020, 9, 947–959. [Google Scholar] [CrossRef]
- Planer, J.D.; Hulverson, M.A.; Arif, J.A.; Ranade, R.M.; Don, R.; Buckner, F.S. Synergy testing of FDA-approved drugs identifies potent drug combinations against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2014, 8, e2977. [Google Scholar] [CrossRef] [PubMed]
- Berenstein, A.J.; Magariños, M.P.; Chernomoretz, A.; Agüero, F. A multilayer network approach for guiding drug repositioning in neglected diseases. PLoS Negl. Trop. Dis. 2016, 10, 1–33. [Google Scholar] [CrossRef]
- Simões-Silva, M.R.; De Araújo, J.S.; Oliveira, G.M.; Demarque, K.C.; Peres, R.B.; D’Almeida-Melo, I.; Batista, D.G.J.; Da Silva, C.F.; Cardoso-Santos, C.; Da Silva, P.B.; et al. Drug repurposing strategy against Trypanosoma cruzi infection: In vitro and in vivo assessment of the activity of metronidazole in mono- and combined therapy. Biochem. Pharmacol. 2017, 145, 46–53. [Google Scholar]
- Ferreira, D.D.; Mesquita, J.T.; Da Costa Silva, T.A.; Romanelli, M.M.; Da Gama Jaen Batista, D.; Da Silva, C.F.; Da Gama, A.N.S.; Neves, B.J.; Melo-Filho, C.C.; Correia Soeiro, M.D.N.; et al. Efficacy of sertraline against Trypanosoma cruzi: An in vitro and in silico study. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 30. [Google Scholar] [CrossRef]
- Kaiser, M.; Mäser, P.; Tadoori, L.P.; Ioset, J.R.; Brun, R.; Sullivan, D.J. Antiprotozoal activity profiling of approved drugs: A starting point toward drug repositioning. PLoS One 2015, 10, e0135556. [Google Scholar] [CrossRef]
- Alonso-Vega, C.; Losada-Galván, I.; Pinazo, M.J.; Sancho Mas, J.; Brustenga, J.G.; Alonso-Padilla, J. The senseless orphanage of Chagas Disease. Expert Opin. Orphan Drugs 2019, 7, 535–545. [Google Scholar] [CrossRef]
- Molina, I.; Goḿez I Prat, J.; Salvador, F.; Treviño, B.; Sulleiro, E.; Serre, N.; Pou, D.; Roure, S.; Cabezos, J.; Valerio, L.; et al. Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 2014, 370, 1899–1908. [Google Scholar] [CrossRef]
- Torrico, F.; Gascon, J.; Ortiz, L.; Alonso-Vega, C.; Pinazo, M.J.; Schijman, A.; Almeida, I.C.; Alves, F.; Strub-Wourgaft, N.; Ribeiro, I.; et al. Treatment of adult chronic indeterminate Chagas disease with benznidazole and three E1224 dosing regimens: A proof-of-concept, randomised, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 419–430. [Google Scholar] [CrossRef]
- Mosca, J.D.; Pitha, P.M. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 1986, 6, 2279–2283. [Google Scholar] [CrossRef]
- Bettiol, E.; Samanovic, M.; Murkin, A.S.; Raper, J.; Buckner, F.; Rodriguez, A. Identification of three classes of heteroaromatic compounds with activity against intracellular Trypanosoma cruzi by chemical library screening. PLoS Negl. Trop. Dis. 2009, 3, e384. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Oh, S.J.; Lee, S.Y.; Im, J.H.; Oh, J.M.; Ryu, C.S.; Kwak, H.C.; Lee, J.Y.; Kang, K.W.; Kim, S.K. HepG2 cells as an in vitro model for evaluation of cytochrome P450 induction by xenobiotics. Arch. Pharm. Res. 2015, 38, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Verlinde, C.L.M.J.; La Flamme, A.C.; Van Voorhis, W.C. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-Galactosidase. Antimicrob. Agents Chemother. 1996, 40, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Reimão, J.Q.; Scotti, M.T.; Tempone, A.G. Anti-leishmanial and anti-trypanosomal activities of 1,4-dihydropyridines: In vitro evaluation and structure-activity relationship study. Bioorganic Med. Chem. 2010, 18, 8044–8053. [Google Scholar] [CrossRef]
- Borges Migliavaca, C.; Stein, C.; Colpani, V.; René Pinto de Sousa Miguel, S.; Nascimento Cruz, L.; Oliveira Dantas, R.; Falavigna, M. Isosorbide and nifedipine for Chagas’ megaesophagus: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2018, 12, 1–13. [Google Scholar] [CrossRef]
- De Rycker, M.; Thomas, J.; Riley, J.; Brough, S.J.; Miles, T.J.; Gray, D.W. Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade. PLoS Negl. Trop. Dis. 2016, 10, e0004584. [Google Scholar] [CrossRef]
- Paolini, E.; Stronati, G.; Guerra, F.; Capucci, A. Flecainide: Electrophysiological properties, clinical indications, and practical aspects. Pharmacol. Res. 2019, 148, 10443. [Google Scholar]
- Quiros, F.R.; Morillo, C.A.; Casas, J.P.; Cubillos, L.A.; Silva, F.A. Charity: Chagas cardiomyopathy bisoprolol intervention study: A randomized double-blind placebo force-titration controlled study with bisoprolol in patients with chronic heart failure secondary to Chagas cardiomyopathy [NCT00323973]. Trials 2006, 7, 21. [Google Scholar] [CrossRef]
- Stein, C.; Castanotto, D.; Krishnan, A.; Nikolaenko, L. Defibrotide (Defitelio): A new addition to the stockpile of food and drug administration-approved oligonucleotide drugs. Molecular Therapy - Nucleic Acids. 2016, 5, e346. [Google Scholar] [CrossRef]
- Landman, G.W.D.; De Bock, G.H.; Van Hateren, K.J.J.; Van Dijk, P.R.; Groenier, K.H.; Gans, R.O.B.; Houweling, S.T.; Bilo, H.J.G.; Kleefstra, N. Safety and efficacy of gliclazide as treatment for type 2 diabetes: A systematic review and meta-analysis of randomized Trials. PLoS One 2014, 9, e82880. [Google Scholar] [CrossRef]
- Penitente, A.R.; Leite, A.L.J.; Costa, G.D.P.; Shrestha, D.; Horta, A.L.; Natali, A.J.; Neves, C.A.; Talvani, A. Enalapril in combination with benznidazole reduces cardiac inflammation and creatine kinases in mice chronically infected with Trypanosoma cruzi. Am. J. Trop. Med. Hyg. 2015, 93, 976–982. [Google Scholar] [CrossRef]
- Loo, C.S.N.; Lam, N.S.K.; Yu, D.; Su, X.Z.; Lu, F. Artemisinin and its derivatives in treating protozoan infections beyond malaria. Pharmacol. Res. 2017, 117, 192–217. [Google Scholar] [CrossRef]
- Moreira, V.R.; De Jesus, L.C.L.; Soares, R.E.P.; Silva, L.D.M.; Pinto, B.A.S.; Melo, M.N.; De AndradePaes, A.M.; Pereira, S.R.F. Meglumine antimoniate (Glucantime) causes oxidative stress-derived DNA damage in Balb/c mice infected by Leishmania (Leishmania) infantum. Antimicrob. Agents Chemother. 2017, 61, e02360-16. [Google Scholar] [CrossRef]
- Andrade, S.G.; Magalhães, L.D.A.; Pessina, D.H. Importance of TNF-α in the course of acute infection with Trypanosoma cruzi: Influence of its inhibition by pentoxifylline treatment. Mem. Inst. Oswaldo Cruz 2008, 103, 21–26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pereira, I.R.; Vilar-Pereira, G.; Moreira, O.C.; Ramos, I.P.; Gibaldi, D.; Britto, C.; Moraes, M.O.; Lannes-Vieira, J. Pentoxifylline reverses chronic experimental chagasic cardiomyopathy in association with repositioning of abnormal CD8+ T-cell response. PLoS Negl. Trop. Dis. 2015, 9, e0003659. [Google Scholar]
- Hobbie, S.N.; Kaiser, M.; Schmidt, S.; Shcherbakov, D.; Janusic, T.; Brun, R.; Böttger, E.C. Genetic reconstruction of protozoan RRNA decoding sites provides a rationale for paromomycin activity against Leishmania and Trypanosoma. PLoS Negl. Trop. Dis. 2011, 5, e1161. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Snowdon, D.; Yardley, V. The activities of four anticancer alkyllysophospholipids against Leishmania donovani, Trypanosoma cruzi and Trypanosoma brucei. J. Antimicrob. Chemother. 1996, 38, 1041–1047. [Google Scholar] [CrossRef]
- Luna, K.P.; Hernandez, I.P.; Rueda, C.M.; Zorro, M.M.; Croft, S.L.; Escobar, P. In vitro susceptibility of Trypanosoma cruzi strains from Santander, Colombia, to hexadecylphosphocholine (Miltefosine), nifurtimox and benznidazole. Biomedica 2009, 29, 448–455. [Google Scholar]
- Santa-Rita, R.M.; Santos Barbosa, H.; Meirelles, M.D.N.S.L.; De Castro, S.L. Effect of the alkyl-lysophospholipids on the proliferation and differentiation of Trypanosoma cruzi. Acta Trop. 2000, 75, 219–228. [Google Scholar]
- Saraiva, V.B.; Gibaldi, D.; Previato, J.O.; Mendonça-Previato, L.; Bozza, M.T.; Freire-de-Lima, C.G.; Heise, N. Proinflammatory and cytotoxic effects of hexadecylphosphocholine (miltefosine) against drug-resistant strains of Trypanosoma cruzi. Antimicrob. Agents Chemother. 2002, 46, 3472–3477. [Google Scholar] [CrossRef]
- Dias, J.C.P.; Schofield, C.J.; Machado, E.M.M.; Fernandes, A.J. Ticks, ivermectin, and experimental Chagas disease. Mem. Inst. Oswaldo Cruz 2005, 100, 829–832. [Google Scholar] [CrossRef][Green Version]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef]
- Flannery, E.L.; Foquet, L.; Chuenchob, V.; Fishbaugher, M.; Billman, Z.; Navarro, M.J.; Betz, W.; Olsen, T.M.; Lee, J.; Camargo, N.; et al. Assessing drug efficacy against Plasmodium falciparum liver stages in vivo. JCI Insight 2018, 3, e92587. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.A.; Weiss, L.M.; Factor, S.; Bilezikian, J.P.; Tanowitz, H.; Wittner, M. Verapamil ameliorates clinical, pathologic and biochemical manifestations of experimental chagasic cardiomyopathy in mice. J. Am. Coll. Cardiol. 1989, 14, 782–789. [Google Scholar] [CrossRef]
- Tanowitz, H.B.; Morris, S.A.; Weiss, L.M.; Bilezikian, J.P.; Factor, S.M.; Wittner, M. Effect of verapamil on the development of chronic experimental Chagas’ disease. Am. J. Trop. Med. Hyg. 1989, 41, 643–649. [Google Scholar] [CrossRef]
- Tanowitz, H.B.; Wittner, M.; Chen, B.; Huang, H.; Weiss, L.M.; Christ, G.J.; Braunstein, V.; Bilezikian, J.P.; Morris, S.A. Effects of verapamil on acute murine Chagas’ disease. J. Parasitol. 1996, 82, 814. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.P.; Tanowitz, H.B.; Chandra, M.; Shtutin, V.; Weiss, L.M.; Morris, S.A.; Factor, S.M.; Huang, H.; Wittner, M.; Shirani, J.; et al. Effects of early and late verapamil administration on the development of cardiomyopathy in experimental chronic Trypanosoma cruzi (Brazil Strain) infection. Parasitol. Res. 2004, 92, 496–501. [Google Scholar]
- Díaz, M.V.; Miranda, M.R.; Campos-Estrada, C.; Reigada, C.; Maya, J.D.; Pereira, C.A.; López-Muñoz, R. Pentamidine exerts in vitro and in vivo anti Trypanosoma cruzi activity and inhibits the polyamine transport in Trypanosoma cruzi. Acta Trop. 2014, 134, 1–9. [Google Scholar] [CrossRef]
- Seguel, V.; Castro, L.; Reigada, C.; Cortes, L.; Díaz, M.V.; Miranda, M.R.; Pereira, C.A.; Lapier, M.; Campos-Estrada, C.; Morello, A.; et al. Pentamidine antagonizes the benznidazole’s effect in vitro, and lacks of synergy in vivo: Implications about the polyamine transport as an anti-Trypanosoma cruzi target. Exp. Parasitol. 2016, 171, 23–32. [Google Scholar] [CrossRef] [PubMed]
- McCabe, R. Primaquine is lethal for intracellular but not extracellular Trypanosoma cruzi. J. Parasitol. 1988, 74, 748–753. [Google Scholar] [CrossRef]
- van der Pluijm, R.W.; Tripura, R.; Hoglund, R.M.; Pyae Phyo, A.; Lek, D.; ul Islam, A.; Anvikar, A.R.; Satpathi, P.; Satpathi, S.; Behera, P.K.; et al. Triple artemisinin-based combination therapies versus artemisinin-based combination therapies for uncomplicated Plasmodium falciparum malaria: A multicentre, open-label, randomised clinical trial. Lancet 2020, 395, 1345–1360. [Google Scholar] [CrossRef]
- Leite, L.R.; Fenelon, G.; Simoes, A.; Silva, G.G.; Friedman, P.A.; De Paola, A.A.V. Clinical usefulness of electrophysiologic testing in patients with ventricular tachycardia and chronic chagasic cardiomyopathy treated with amiodarone or sotalol. J. Cardiovasc. Electrophysiol. 2003, 14, 567–573. [Google Scholar]
- Andriani, G.; Chessler, A.-D.C.; Courtemanche, G.; Burleigh, B.A.; Rodriguez, A. Activity in vivo of anti-Trypanosoma cruzi compounds selected from a high throughput screening. PLoS Negl. Trop. Dis. 2011, 5, e1298. [Google Scholar] [CrossRef]
- Martinez-Peinado, N.; Cortes-Serra, N.; Torras-Claveria, L.; Pinazo, M.-J.; Gascon, J.; Bastida, J.; Alonso-Padilla, J. Amaryllidaceae alkaloids with anti-Trypanosoma cruzi activity. Parasit. Vectors 2020, 13, 299. [Google Scholar] [PubMed]
- Martinez-Peinado, N.; Martori, C.; Cortes-Serra, N.; Sherman, J.; Rodriguez, A.; Gascon, J.; Alberola, J.; Pinazo, M.-J.; Rodriguez-Cortes, A.; Alonso-Padilla, J. Anti-Trypanosoma cruzi activity of metabolism modifier compounds. Int. J. Mol. Sci. 2021, 22, 688. [Google Scholar] [CrossRef]
- Alonso-Padilla, J.; Cotillo, I.; Presa, J.L.; Cantizani, J.; Peña, I.; Bardera, A.I.; Martín, J.J.; Rodriguez, A. Automated high-content assay for compounds selectively toxic to Trypanosoma cruzi in a myoblastic cell line. PLoS Negl. Trop. Dis. 2015, 9, e0003493. [Google Scholar] [CrossRef]
- Franco, C.H.; Alcântara, L.M.; Chatelain, E.; Freitas-Junior, L.; Moraes, C.B. Drug discovery for Chagas disease: Impact of different host cell lines on assay performance and hit compound selection. Trop. Med. Infect. Dis. 2019, 4, 82. [Google Scholar]
- Mitchel, F.L. The implementation of quality control and factors affecting its success. Ann. Clin. Biochem. 1969, 6, 119–122. [Google Scholar] [CrossRef]
- Anti-Infectives Screening Core Services | NYU Langone Health. Available online: https://med.nyu.edu/research/scientific-cores-shared-resources/anti-infectives-screening-core/services (accessed on 28 January 2021).
- Crouch, S.P.M.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar]
- Francisco, A.F.; Jayawardhana, S.; Lewis, M.D.; Taylor, M.C.; Kelly, J.M. Biological factors that impinge on Chagas disease drug development. Parasitology 2017, 144, 1871–1880. [Google Scholar] [CrossRef]
- Lewis, M.D.; Francisco, A.F.; Taylor, M.C.; Kelly, J.M. A new experimental model for assessing drug efficacy against Trypanosoma cruzi infection based on highly sensitive in vivo imaging. J. Biomol. Screen. 2015, 20, 36–43. [Google Scholar]
- Timm, B.L.; Da Silva, P.B.; Batista, M.M.; Farahat, A.A.; Kumar, A.; Boykin, D.W.; Soeiro, M.N.C. In vitro investigation of the efficacy of novel diamidines against Trypanosoma cruzi. Parasitology 2014, 141, 1272–1276. [Google Scholar] [CrossRef]
- Wispelwey, B.; Pearson, R. Pentamidine: A risk-benefit analysis. Drug Saf. 1990, 5, 212–219. [Google Scholar] [CrossRef]
- Lewis, M.D.; Francisco, A.F.; Taylor, M.C.; Jayawardhana, S.; Kelly, J.M. Host and parasite genetics shape a link between Trypanosoma cruzi infection dynamics and chronic cardiomyopathy. Cell Microbiol. 2016, 18, 1429–1443. [Google Scholar] [CrossRef]
- Sunyoto, T.; Potet, J.; Boelaert, M. Why miltefosine—A life-saving drug for leishmaniasis—Is unavailable to people who need it the most. BMJ Glob. Health 2018, 3, e000709. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Martinez, A.K.; Rodriguez-Durán, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of action of miltefosine on Leishmania donovani involves the impairment of acidocalcisome function and the activation of the sphingosine-dependent plasma membrane Ca2+ channel. Antimicrob. Agents Chemother. 2018, 62, e01614-17. [Google Scholar] [CrossRef]
- Rohloff, P.; Montalvetti, A.; Docampo, R. Acidocalcisomes and the contractile vacuole complex are involved in osmoregulation in Trypanosoma cruzi. J. Biol. Chem. 2004, 279, 52270–52281. [Google Scholar]
- Benaim, G.; Paniz-Mondolfi, A.E.; Sordillo, E.M.; Martinez-Sotillo, N. Disruption of intracellular calcium homeostasis as a therapeutic target against Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2020, 10, 46. [Google Scholar] [CrossRef]
- Braga, S.S. Multi-target drugs active against leishmaniasis: A paradigm of drug repurposing. Eur. J. Med. Chem. 2019, 183, 111660. [Google Scholar] [CrossRef]
- Sundar, S.; Olliaro, P.L. Miltefosine in the treatment of leishmaniasis: Clinical evidence for informed clinical risk management. Ther. Clin. Risk Manag. 2007, 3, 733–740. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trade Name | Active Ingredient | Class | Route of Administration | Molecular Weight (g/mol) | References |
|---|---|---|---|---|---|
| Adalat | nifedipine | calcium channel blocker | oral | 346.34 | [26,27] |
| Amlodipine Normon | amlodipine | calcium channel blocker | oral | 408.88 | [14,26,28] |
| Apocard | flecainide | antiarrhythmic | oral | 414.34 | [29] |
| Atenolol Normon | atenolol | beta blocker | oral | 266.34 | [14] |
| Biocoryl | procainamide | antiarrhythmic | oral | 235.33 | [14] |
| Bisoprolol Normon | bisoprolol | beta blocker | oral | 325.44 | [30] |
| Daraprim | pyrimethamine | antiprotozoal | oral | 248.71 | [14] |
| Defitelio | defibrotide | antithrombotic | intravenous | 444.40 | [31] |
| Diamicron | gliclazide | antidiabetic | oral | 323.41 | [32] |
| Enalapril Normon | enalapril | ACE inhibitor | oral | 376.45 | [14,33] |
| Eskazole | albendazole | antihelminthic and antiprotozoal | oral | 256.33 | [14] |
| Eurartesim | piperaquine tetraphosphate-dihydroartemisinin | antimalarial | oral | 927.49–284.35 | [34] |
| Glucantime | meglumine antimoniate | antileishmanial | intramuscular | 365.98 | [26,35] |
| Glucophage | metformin hydrochloride | antidiabetic | oral | 129.16 | [14] |
| Hemovas | pentoxifylline | hemorrheologic agent | oral | 278.31 | [14,36,37] |
| Humatin | paramomycin | antimicrobial | oral | 615.63 | [38] |
| Impavido | miltefosine | antiprotozoal | oral | 407.57 | [39,40,41,42] |
| Iver P (ELEA) | ivermectin | antihelminthic | oral | 875.10 | [43] |
| Quinine sulfate (Hospital Clinic) | quinine sulfate | antimalarial | oral | 782.96 | [44] |
| Lidocaine Braun | lidocaine | local anesthetic | intravenous | 234.34 | [14] |
| Lomper | mebendazole | antihelminthic | oral | 295.29 | [14] |
| Malarone | atovaquone-proguanil | antimalarial | oral | 366.84 | [45] |
| Manidon | verapamil | calcium channel blocker | oral/intravenous | 454.60 | [46,47,48,49] |
| Masdil | diltiazem hydrochloride | calcium channel blocker | oral | 414.52 | [14] |
| Menaderm Otológico | beclometasone dipropionate-clioquinol | fungal/antibacterial | otic | 521.04–304.91 | - |
| Nerdipina | nicardipine | calcium channel blocker | oral | 479.53 | [14,26] |
| Pentacarinat | pentamidine | antiprotozoal | intravenous/intramuscular/inhalation | 340.42 | [14,26,50,51] |
| Primaquine (Hospital Clinic) | primaquine | antimalarial | oral | 259.35 | [14,52] |
| Riamet | artemether-lumefrantine | antimalarial | oral | 298.37–528.94 | [53] |
| Solgol | nadolol | beta blocker | oral | 309.40 | [14] |
| Sotapor | sotalol | beta blocker | oral | 272.36 | [54] |
| Tricolam | tinidazole | antiprotozoal | oral | 247.27 | [14] |
| Drug | Vero Cells Assays | NIH-3T3 Cells Assays | HepG2 Assay | Anti-Amastigote Assay | |||||
|---|---|---|---|---|---|---|---|---|---|
| [IC50 (SD)] µmol L−1 | [TC50 (SD)] µmol L−1 | SI | [IC50 (SD)] µmol L−1 | [TC50 (SD)] µmol L−1 | SI | [TC50 (SD)] µmol L−1 | [IC50 (SD)] µmol L−1 | SI | |
| Benznidazole | 1.93 (0.82) | 242.2 (13.93) | 125.5 | - | - | - | 229.8 (18.54) | 2.66 (0.14) | 91.1 |
| Atovaquone-proguanil | 1.26 (0.14) | 27.13 (5.05) | 21.5 | 1.32 (0.07) | >50 | >50 | 34.36 (5.88) | 1.85 (0.06) | 14.7 |
| Miltefosine | 0.018 (0.0015) | 78.99 (10.55) | 4388.3 | 0.037 (0.001) | 1.95 (0.57) | 52.7 | 51.28 (7.51) | 1.25 (0.05) | 63.2 |
| Lidocaine # | 0.016 (0.0015) | 0.23 (0.027) | 14.4 | - | - | - | - | - | - |
| Nifedipine | 0.19 (0.018) | 1.97 (0.267) | 10.4 | - | - | - | - | - | - |
| Pentamidine | 1.01 (0.55) | 78.96 (15.55) | 78.2 | 0.13 (0.005) | 5.9 (0.15) | 45.4 | 39.4 (5.20) | - | - |
| Piperaquine tetraphosphate-dihydroartemisinin | 3.95 (0.51) | 75.27 (16.56) | 19.1 | 4.05 (0.72) | 27.33 (3.68) | 6.8 | - | - | - |
| Verapamil | 3.44 (0.44) | 197.4 (25.54) | 57.4 | 0.60 (0.04) | 8.16 (1.63) | 13.6 | 170.5 (13.14) | 122.5 (7.04) | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Peinado, N.; Cortes-Serra, N.; Sherman, J.; Rodriguez, A.; Bustamante, J.M.; Gascon, J.; Pinazo, M.-J.; Alonso-Padilla, J. Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs. Microorganisms 2021, 9, 406. https://doi.org/10.3390/microorganisms9020406
Martinez-Peinado N, Cortes-Serra N, Sherman J, Rodriguez A, Bustamante JM, Gascon J, Pinazo M-J, Alonso-Padilla J. Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs. Microorganisms. 2021; 9(2):406. https://doi.org/10.3390/microorganisms9020406
Chicago/Turabian StyleMartinez-Peinado, Nieves, Nuria Cortes-Serra, Julian Sherman, Ana Rodriguez, Juan M. Bustamante, Joaquim Gascon, Maria-Jesus Pinazo, and Julio Alonso-Padilla. 2021. "Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs" Microorganisms 9, no. 2: 406. https://doi.org/10.3390/microorganisms9020406
APA StyleMartinez-Peinado, N., Cortes-Serra, N., Sherman, J., Rodriguez, A., Bustamante, J. M., Gascon, J., Pinazo, M.-J., & Alonso-Padilla, J. (2021). Identification of Trypanosoma cruzi Growth Inhibitors with Activity In Vivo within a Collection of Licensed Drugs. Microorganisms, 9(2), 406. https://doi.org/10.3390/microorganisms9020406