
Global Transcriptional Response of Methylorubrum extorquens to Formaldehyde Stress Expands the Role of EfgA and Is Distinct from Antibiotic Translational Inhibition

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Media, and Chemicals
2.2. Growth Quantitation
2.3. Formaldehyde Quantification
2.4. Transcriptomic Analysis
2.4.1. Cell Treatment
2.4.2. Harvesting Cells
2.4.3. RNA Sequencing
2.4.4. Data Analysis
2.5. Gene Enrichment
3. Results
3.1. Formaldehyde Stress Induces a Quiescent State
3.2. Cells Resume Growth When the Formaldehyde Stress Is Reduced
3.3. Initial Experiments to Identify Timescale of Response for Further Transcriptomic Analyses
3.4. Overall Pattern of Gene Expression Changes Revealed via Principal Component Analysis
3.5. efgA Genotype Does Not Impact the Transcriptome during Growth upon Succinate in the Absence of Formaldehyde
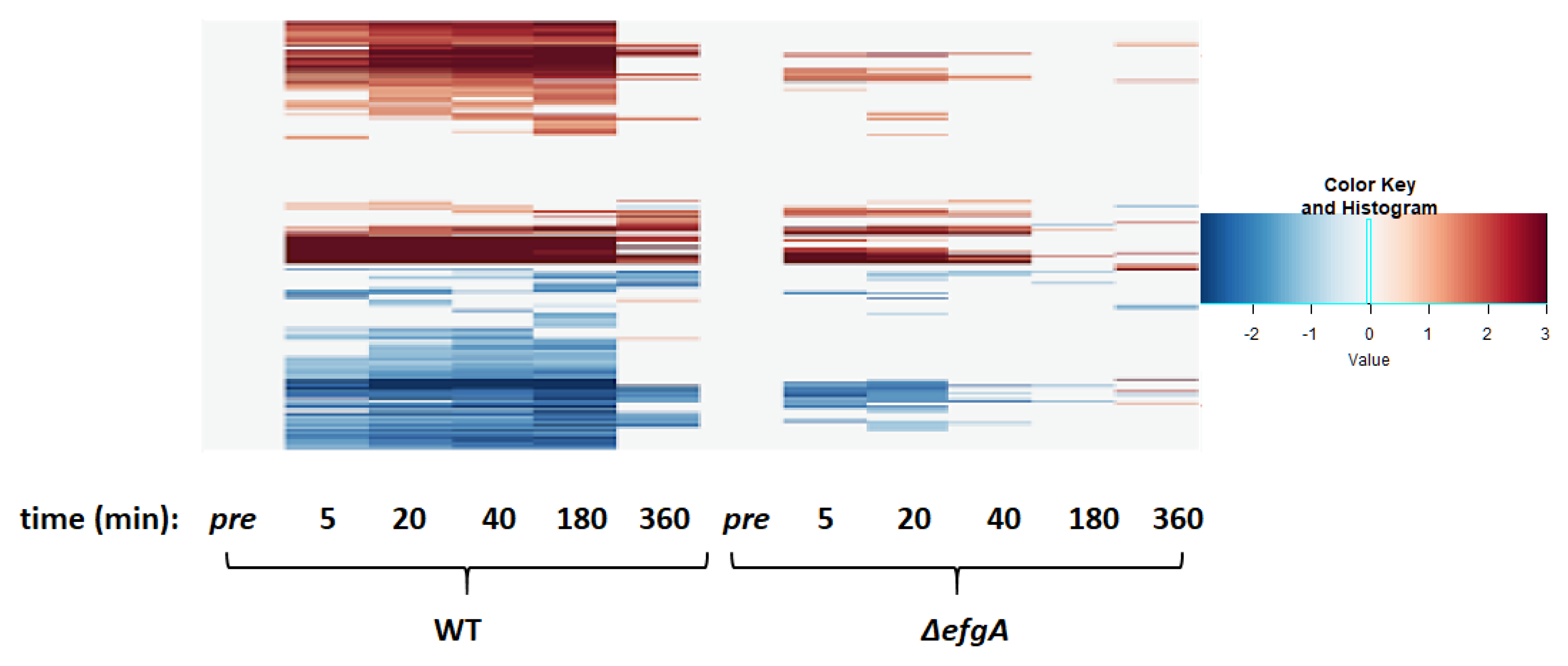
3.6. The Global Response to Formaldehyde Is Extensive and Predominantly Mediated by Rapid Action of EfgA
3.7. Loss of Viability Precedes the Transcriptomic Response to Kanamycin
3.8. Response to Kanamycin and Formaldehyde Involve Shared Pathways
3.9. Response of Methylotrophy Genes
3.9.1. Methanol Oxidation
3.9.2. Formaldehyde Oxidation
3.9.3. Formate Branchpoint
3.9.4. Serine Cycle
3.10. Wild-Type Response to Formaldehyde
3.11. Investigating Known Modulators of Formaldehyde Tolerance
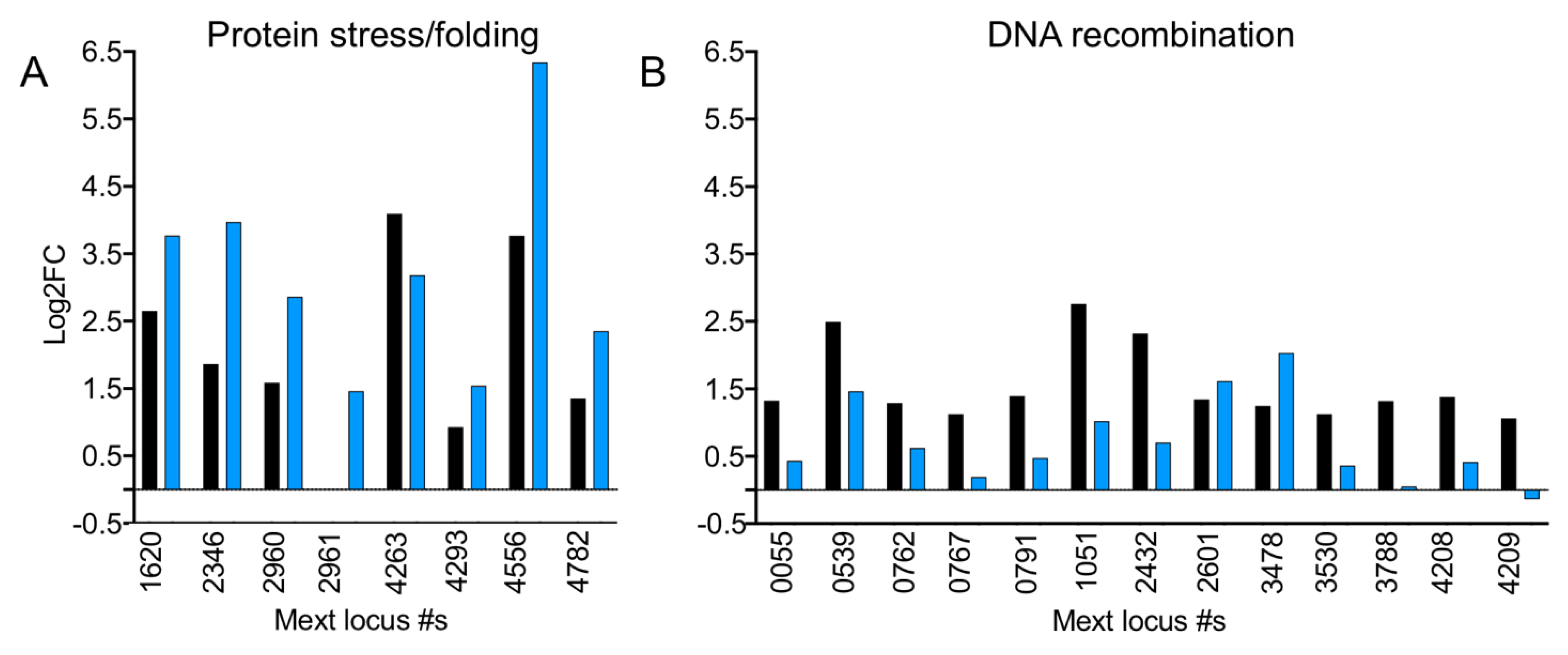
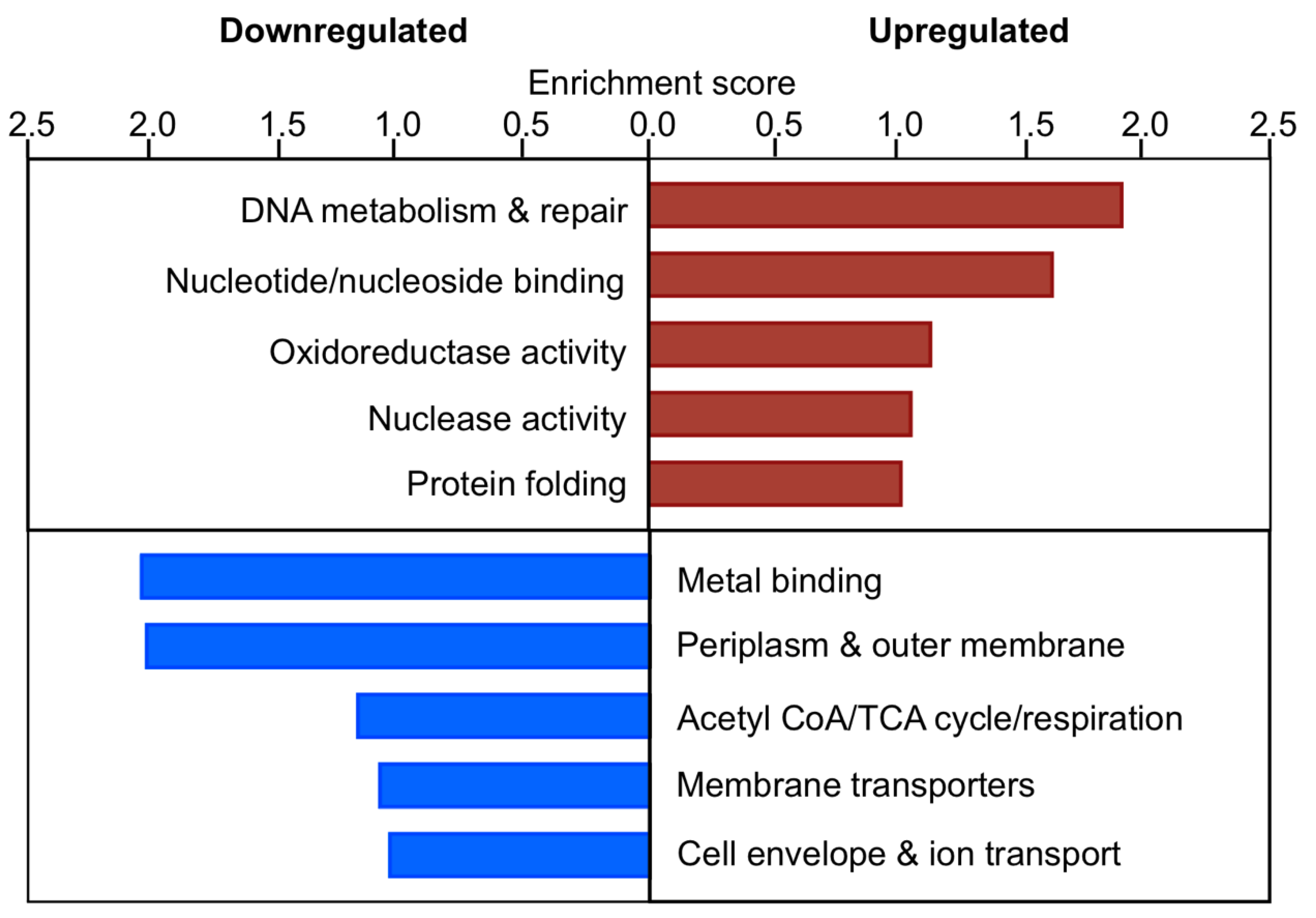
3.12. In the Absence of EfgA Cells Have Increased Response to Proteotoxicity and Genotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohanski, M.A.; DePristo, M.A.; Collins, J.J. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 2010, 37, 311–320. [Google Scholar] [CrossRef]
- Tamás, M.J.; Sharma, S.K.; Ibstedt, S.; Jacobson, T.; Christen, P. Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 2014, 4, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef]
- Ireland, H.E.; Elyse Ireland, H.; Harding, S.J.; Bonwick, G.A.; Jones, M.; Smith, C.J.; Williams, J.H.H. Evaluation of heat shock protein 70 as a biomarker of environmental stress in Fucus serratus and Lemna minor. Biomarkers 2004, 9, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef]
- Hoffman, E.A.; Frey, B.L.; Smith, L.M.; Auble, D.T. Formaldehyde crosslinking: A tool for the study of chromatin complexes. J. Biol. Chem. 2015, 290, 26404–26411. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Matsuda, T.; Yagi, T. Genotoxicity of formaldehyde: Molecular basis of DNA damage and mutation. Front. Environ. Sci. Eng. China 2014, 2, 36. [Google Scholar] [CrossRef]
- Zhang, H.; Xiong, Y.; Chen, J. DNA-protein cross-link repair: What do we know now? Cell Biosci. 2020, 10, 3. [Google Scholar] [CrossRef]
- Xie, M.-Z.; Shoulkamy, M.I.; Salem, A.M.H.; Oba, S.; Goda, M.; Nakano, T.; Ide, H. Aldehydes with high and low toxicities inactivate cells by damaging distinct cellular targets. Mutat. Res. 2016, 786, 41–51. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T. Molecular mechanisms of aldehyde toxicity: A chemical perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef]
- Chen, N.H.; Djoko, K.Y.; Veyrier, F.J.; McEwan, A.G. Formaldehyde stress responses in bacterial pathogens. Front. Microbiol. 2016, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Achkor, H.; Díaz, M.; Fernández, M.R.; Biosca, J.A.; Parés, X.; Martínez, M.C. Enhanced formaldehyde detoxification by overexpression of glutathione-dependent formaldehyde dehydrogenase from Arabidopsis. Plant Physiol. 2003, 132, 2248–2255. [Google Scholar] [CrossRef]
- Burgos-Barragan, G.; Wit, N.; Meiser, J.; Dingler, F.A.; Pietzke, M.; Mulderrig, L.; Pontel, L.B.; Rosado, I.V.; Brewer, T.F.; Cordell, R.L.; et al. Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 2017, 548, 549–554. [Google Scholar] [CrossRef]
- Giese, M.; Bauer-Doranth, U.; Langebartels, C.; Sandermann, H., Jr. Detoxification of formaldehyde by the spider plant (Chlorophytum comosum L.) and by Soybean (Glycine max L.) cell-suspension cultures. Plant Physiol. 1994, 104, 1301–1309. [Google Scholar] [CrossRef]
- Harms, N.; Ras, J.; Reijnders, W.N.; van Spanning, R.J.; Stouthamer, A.H. S-formylglutathione hydrolase of Paracoccus denitrificans is homologous to human esterase D: A universal pathway for formaldehyde detoxification? J. Bacteriol. 1996, 178, 6296–6299. [Google Scholar] [CrossRef]
- Höög, J.-O.; Hedberg, J.J.; Strömberg, P.; Svensson, S. Mammalian alcohol dehydrogenase—Functional and structural implications. J. Biomed. Sci. 2001, 8, 71–76. [Google Scholar] [CrossRef]
- Lee, B.; Yurimoto, H.; Sakai, Y.; Kato, N. Physiological role of the glutathione-dependent formaldehyde dehydrogenase in the methylotrophic yeast Candida Boidinii. Microbiology 2002, 148, 2697–2704. [Google Scholar] [CrossRef]
- Sanghani, P.C.; Robinson, H.; Bosron, W.F.; Hurley, T.D. Human glutathione-dependent formaldehyde dehydrogenase. Structures of apo, binary, and inhibitory ternary complexes. Biochemistry 2002, 41, 10778–10786. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A. Cofactor-dependent pathways of formaldehyde oxidation in methylotrophic bacteria. Arch. Microbiol. 2002, 178, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E. Tetrahydrofolate and tetrahydromethanopterin compared: Functionally distinct carriers in C1 metabolism. Biochem. J. 2000, 350 Pt 3, 609–629. [Google Scholar] [CrossRef]
- Kato, N.; Yurimoto, H.; Thauer, R.K. The physiological role of the ribulose monophosphate pathway in bacteria and archaea. Biosci. Biotechnol. Biochem. 2006, 70, 10–21. [Google Scholar] [CrossRef]
- Marx, C.J.; Miller, J.A.; Chistoserdova, L.; Lidstrom, M.E. Multiple formaldehyde oxidation/detoxification pathways in Burkholderia fungorum LB400. J. Bacteriol. 2004, 186, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Vorholt, J.A.; Marx, C.J.; Lidstrom, M.E.; Thauer, R.K. Novel formaldehyde-activating enzyme in Methylobacterium extorquens AM1 required for growth on methanol. J. Bacteriol. 2000, 182, 6645–6650. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Chistoserdova, L.; Lidstrom, M.E. Formaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J. Bacteriol. 2003, 185, 7160–7168. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Van Dien, S.J.; Lidstrom, M.E. Flux analysis uncovers key role of functional redundancy in formaldehyde metabolism. PLoS Biol. 2005, 3, e16. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gomez, N.C.; Good, N.M.; Lidstrom, M.E. Methenyl-Dephosphotetrahydromethanopterin is a regulatory signal for acclimation to changes in substrate availability in Methylobacterium extorquens AM1. J. Bacteriol. 2015, 197, 2020–2026. [Google Scholar] [CrossRef]
- Bazurto, J.V.; Nayak, D.D.; Ticak, T.; Davlieva, M.; Lee, J.A.; Lambert, L.B.; Benski, O.J.; Quates, C.J.; Johnson, J.L.; Patel, J.S.; et al. EfgA is a conserved formaldehyde sensor that halts bacterial translation in response to elevated formaldehyde. BioRxiv 2020, 343392. [Google Scholar] [CrossRef]
- Marx, C.J.; Bringel, F.; Chistoserdova, L.; Moulin, L.; Farhan Ul Haque, M.; Fleischman, D.E.; Gruffaz, C.; Jourand, P.; Knief, C.; Lee, M.-C.; et al. Complete genome sequences of six strains of the genus Methylobacterium. J. Bacteriol. 2012, 194, 4746–4748. [Google Scholar] [CrossRef]
- Knief, C.; Frances, L.; Vorholt, J.A. Competitiveness of diverse Methylobacterium strains in the phyllosphere of Arabidopsis thaliana and identification of representative models, including M. extorquens PA1. Microb. Ecol. 2010, 60, 440–452. [Google Scholar] [CrossRef]
- Delaney, N.F.; Kaczmarek, M.E.; Ward, L.M.; Swanson, P.K.; Lee, M.-C.; Marx, C.J. Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS ONE 2013, 8, e62957. [Google Scholar] [CrossRef]
- Nash, T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J. 1953, 55, 416–421. [Google Scholar] [CrossRef]
- Stead, M.B.; Agrawal, A.; Bowden, K.E.; Nasir, R.; Mohanty, B.K.; Meagher, R.B.; Kushner, S.R. RNA snap TM: A rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 2012, 40, e156. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. gplots: Various R programming tools for plotting data. R Package Version 2009, 2, 1. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef]
- Fruzangohar, M.; Ebrahimie, E.; Adelson, D.L. A novel hypothesis-unbiased method for Gene Ontology enrichment based on transcriptome data. PLoS ONE 2017, 12, e0170486. [Google Scholar] [CrossRef]
- Fruzangohar, M.; Ebrahimie, E.; Ogunniyi, A.D.; Mahdi, L.K.; Paton, J.C.; Adelson, D.L. Comparative GO: A web application for comparative gene ontology and gene ontology-based gene selection in bacteria. PLoS ONE 2013, 8, e58759. [Google Scholar] [CrossRef]
- Rittershaus, E.S.C.; Baek, S.-H.; Sassetti, C.M. The normalcy of dormancy: Common themes in microbial quiescence. Cell Host Microbe 2013, 13, 643–651. [Google Scholar] [CrossRef]
- Bollenbach, T.; Quan, S.; Chait, R.; Kishony, R. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell 2009, 139, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, M.; Ershov, D.; Rozendaal, R.; Walker, N.; Schultz, D.; Kishony, R.; Levin, P.A.; Tans, S.J. Size laws and division ring dynamics in filamentous Escherichia coli cells. Curr. Biol. 2018, 28, 972–979.e5. [Google Scholar] [CrossRef]
- Imlay, J.A.; Linn, S. Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide. J. Bacteriol. 1987, 169, 2967–2976. [Google Scholar] [CrossRef]
- Peyraud, R.; Kiefer, P.; Christen, P.; Portais, J.-C.; Vorholt, J.A. Co-consumption of methanol and succinate by Methylobacterium extorquens AM1. PLoS ONE 2012, 7, e48271. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Kosály, G.; Lidstrom, M.E. Formate as the main branch point for methylotrophic metabolism in Methylobacterium extorquens AM1. J. Bacteriol. 2008, 190, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Riazi, S.; Nemati, S.; Bazurto, J.V.; Vasdekis, A.E.; Ridenhour, B.J.; Remien, C.H.; Marx, C.J. Microbial phenotypic heterogeneity in response to a metabolic toxin: Continuous, dynamically shifting distribution of formaldehyde tolerance in Methylobacterium extorquens populations. PLoS Genet. 2019, 15, e1008458. [Google Scholar] [CrossRef] [PubMed]
- Bazurto, J.V.; Bruger, E.L.; Lee, J.A.; Lambert, L.B.; Marx, C.J. Formaldehyde-responsive proteins, TtmR and EfgA, reveal a tradeoff between formaldehyde resistance and efficient transition to methylotrophy in Methylorubrum extorquens. BioRxiv 2020, 346494. [Google Scholar] [CrossRef]
- O’Donovan, M.R.; Mee, C.D. Formaldehyde is a bacterial mutagen in a range of Salmonella and Escherichia indicator strains. Mutagenesis 1993, 8, 577–581. [Google Scholar] [CrossRef]
- Ashby, J.; Callander, R.D.; Rose, F.L. Weak mutagenicity to Salmonella of the formaldehyde-releasing anti-tumour agent hexamethylmelamine. Mutat. Res. 1985, 142, 121–125. [Google Scholar] [CrossRef]
- Takahashi, K.; Morita, T.; Kawazoe, Y. Mutagenic characteristics of formaldehyde on bacterial systems. Mutat. Res. 1985, 156, 153–161. [Google Scholar] [CrossRef]
- Nishioka, H. Others Lethal and mutagenic action of formaldehyde in Hcr+ and Hcr− strains of Escherichia coli. Mutat. Res. 1973, 17, 261–265. [Google Scholar] [CrossRef]
- Temcharoen, P.; Thilly, W.G. Toxic and mutagenic effects of formaldehyde in Salmonella typhimurium. Mutat. Res. 1983, 119, 89–93. [Google Scholar] [CrossRef]
- De Graaf, B.; Clore, A.; McCullough, A.K. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair. 2009, 8, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katafuchi, A.; Matsubara, M.; Terato, H.; Tsuboi, T.; Masuda, T.; Tatsumoto, T.; Pack, S.P.; Makino, K.; Croteau, D.L.; et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009, 284, 27065–27076. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Takahashi, A.; Kondo, N.; Mori, E.; Okamoto, N.; Nakagawa, Y.; Ohnishi, K.; Zdzienicka, M.Z.; Thompson, L.H.; Helleday, T.; et al. Repair pathways independent of the Fanconi anemia nuclear core complex play a predominant role in mitigating formaldehyde-induced DNA damage. Biochem. Biophys. Res. Commun. 2011, 404, 206–210. [Google Scholar] [CrossRef]
- Ridpath, J.R.; Nakamura, A.; Tano, K.; Luke, A.M.; Sonoda, E.; Arakawa, H.; Buerstedde, J.-M.; Gillespie, D.A.F.; Sale, J.E.; Yamazoe, M.; et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007, 67, 11117–11122. [Google Scholar] [CrossRef]
- Rosado, I.V.; Langevin, F.; Crossan, G.P.; Takata, M.; Patel, K.J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 2011, 18, 1432–1434. [Google Scholar] [CrossRef] [PubMed]
- Pitten, F.A.; Kramer, A.; Herrmann, K.; Bremer, J.; Koch, S. Formaldehyde neurotoxicity in animal experiments. Pathol. Res. Pract. 2000, 196, 193–198. [Google Scholar] [CrossRef]
- Reddy, N.J.; Sudini, M.; Lewis, L.D. Delayed neurological sequelae from ethylene glycol, diethylene glycol and methanol poisonings. Clin. Toxicol. 2010, 48, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Atienza, S.; Rubis, B.; McCarthy, C.; Zhitkovich, A. Formaldehyde is a potent proteotoxic stressor causing rapid Heat Shock Transcription Factor 1 activation and Lys48-linked polyubiquitination of proteins. Am. J. Pathol. 2016, 186, 2857–2868. [Google Scholar] [CrossRef]
- Foster, P.L. Stress responses and genetic variation in bacteria. Mutat. Res. 2005, 569, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, C.; Konan, K.V.; Sarsero, J.P. Some novel transcription attenuation mechanisms used by bacteria. Biochimie 1996, 78, 1017–1024. [Google Scholar] [CrossRef]
- Gollnick, P.; Babitzke, P. Transcription attenuation. Biochim. Biophys. Acta 2002, 1577, 240–250. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazurto, J.V.; Riazi, S.; D’Alton, S.; Deatherage, D.E.; Bruger, E.L.; Barrick, J.E.; Marx, C.J. Global Transcriptional Response of Methylorubrum extorquens to Formaldehyde Stress Expands the Role of EfgA and Is Distinct from Antibiotic Translational Inhibition. Microorganisms 2021, 9, 347. https://doi.org/10.3390/microorganisms9020347
Bazurto JV, Riazi S, D’Alton S, Deatherage DE, Bruger EL, Barrick JE, Marx CJ. Global Transcriptional Response of Methylorubrum extorquens to Formaldehyde Stress Expands the Role of EfgA and Is Distinct from Antibiotic Translational Inhibition. Microorganisms. 2021; 9(2):347. https://doi.org/10.3390/microorganisms9020347
Chicago/Turabian StyleBazurto, Jannell V., Siavash Riazi, Simon D’Alton, Daniel E. Deatherage, Eric L. Bruger, Jeffrey E. Barrick, and Christopher J. Marx. 2021. "Global Transcriptional Response of Methylorubrum extorquens to Formaldehyde Stress Expands the Role of EfgA and Is Distinct from Antibiotic Translational Inhibition" Microorganisms 9, no. 2: 347. https://doi.org/10.3390/microorganisms9020347
APA StyleBazurto, J. V., Riazi, S., D’Alton, S., Deatherage, D. E., Bruger, E. L., Barrick, J. E., & Marx, C. J. (2021). Global Transcriptional Response of Methylorubrum extorquens to Formaldehyde Stress Expands the Role of EfgA and Is Distinct from Antibiotic Translational Inhibition. Microorganisms, 9(2), 347. https://doi.org/10.3390/microorganisms9020347