
Cross-Feeding of a Toxic Metabolite in a Synthetic Lignocellulose-Degrading Microbial Community

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
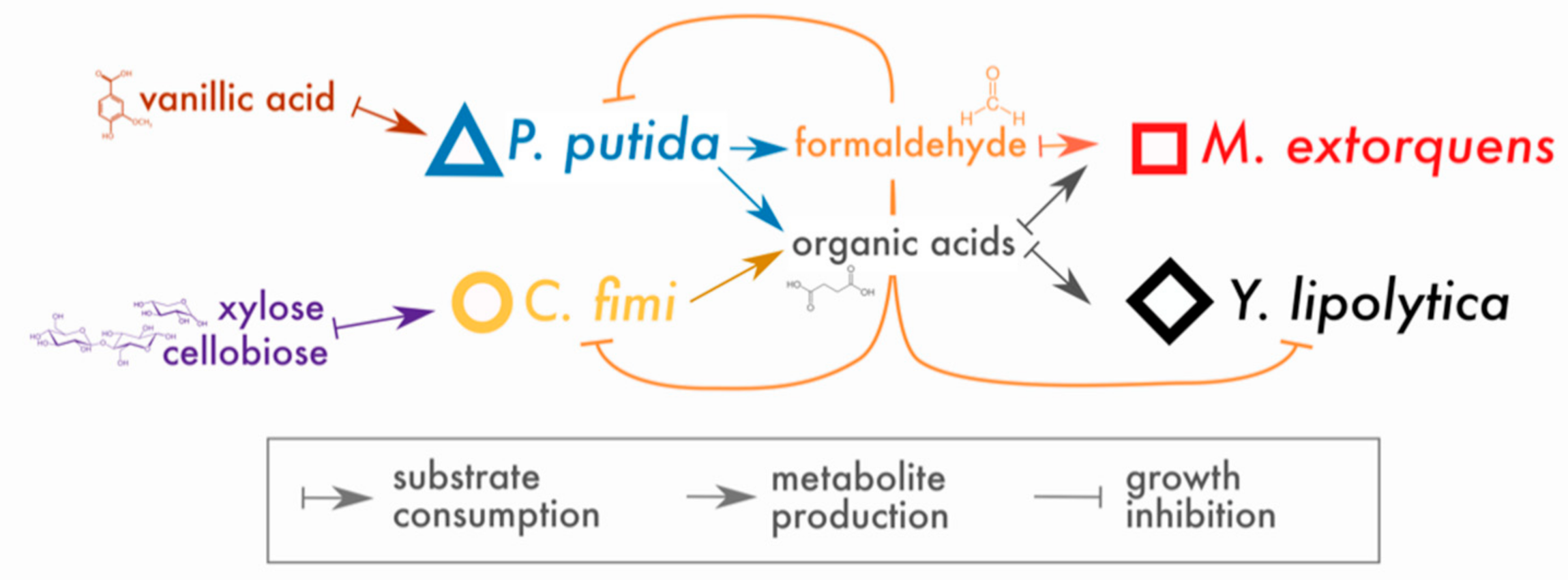
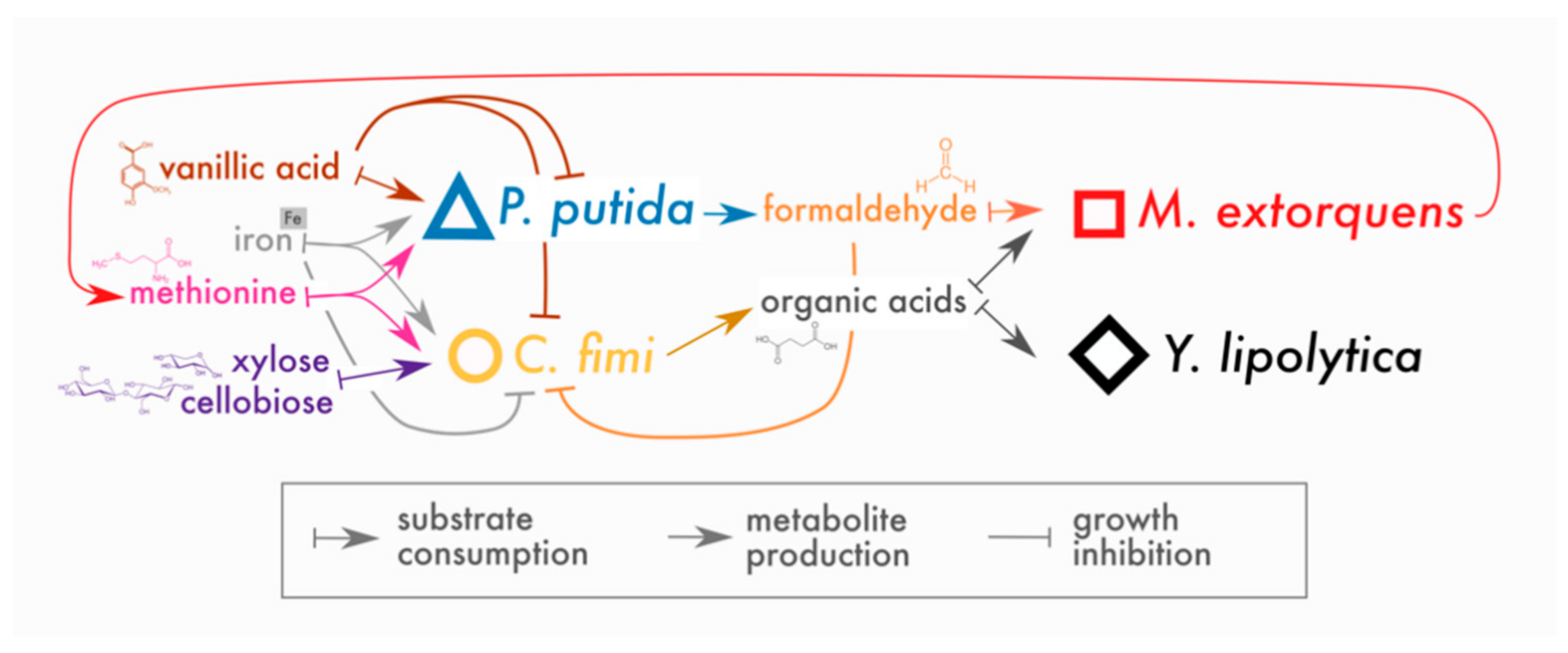
3. Results
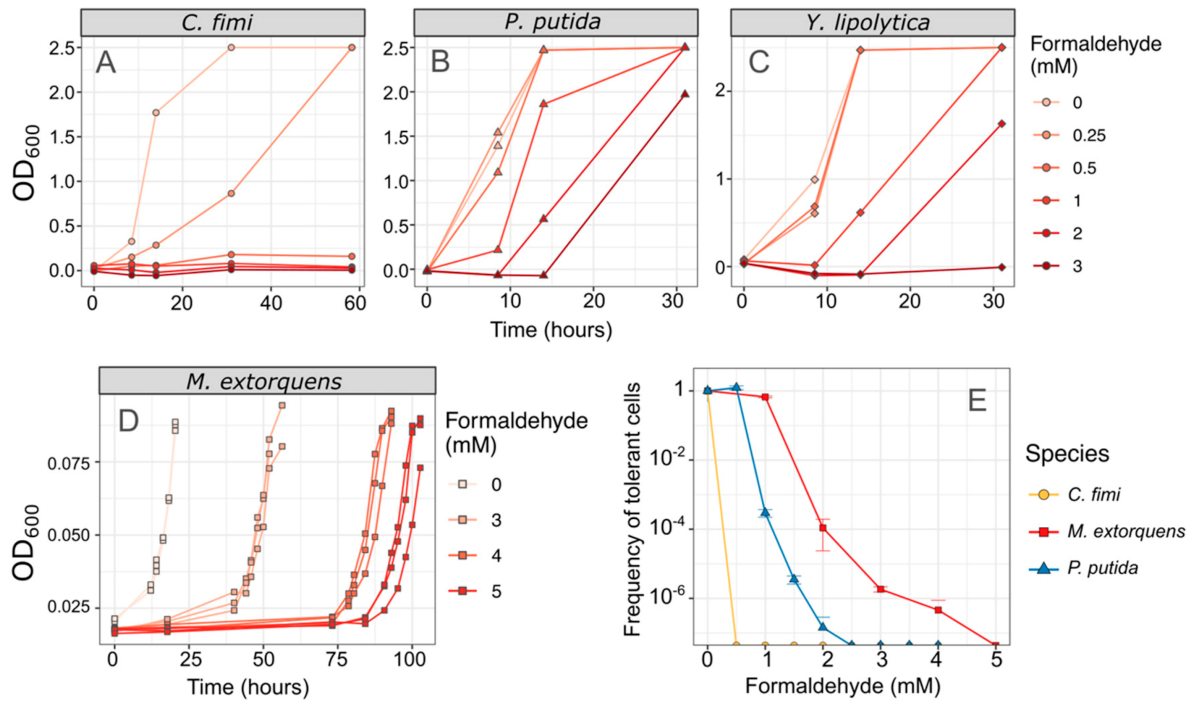
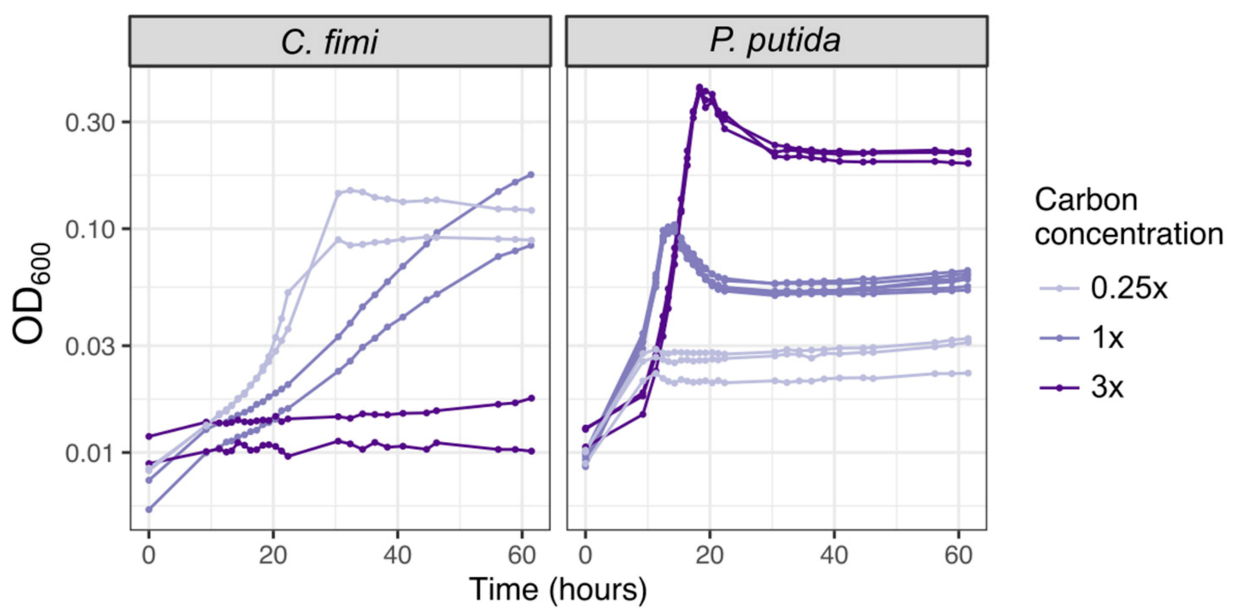
3.1. Both Vanillic Acid and the Formaldehyde Generated during Its Degradation Are Toxic to Consortium Members
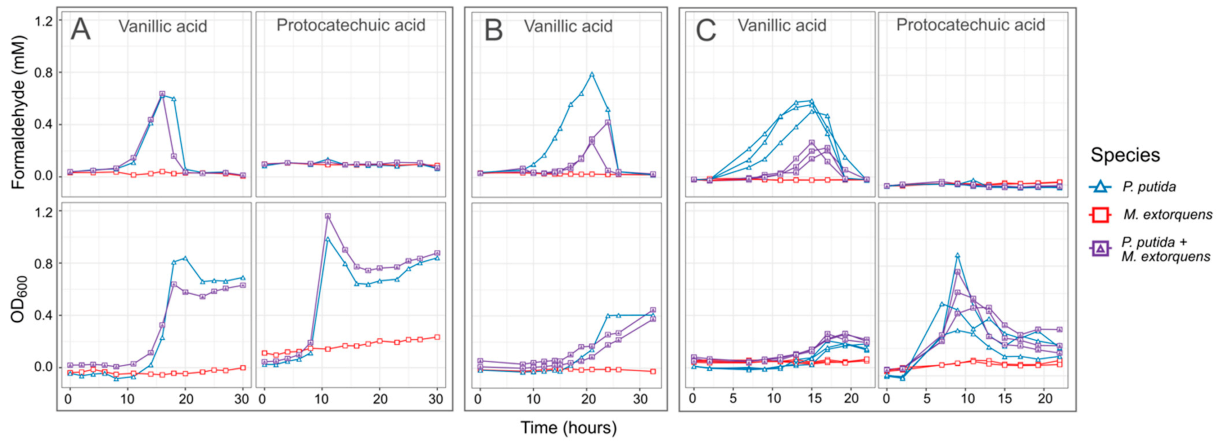
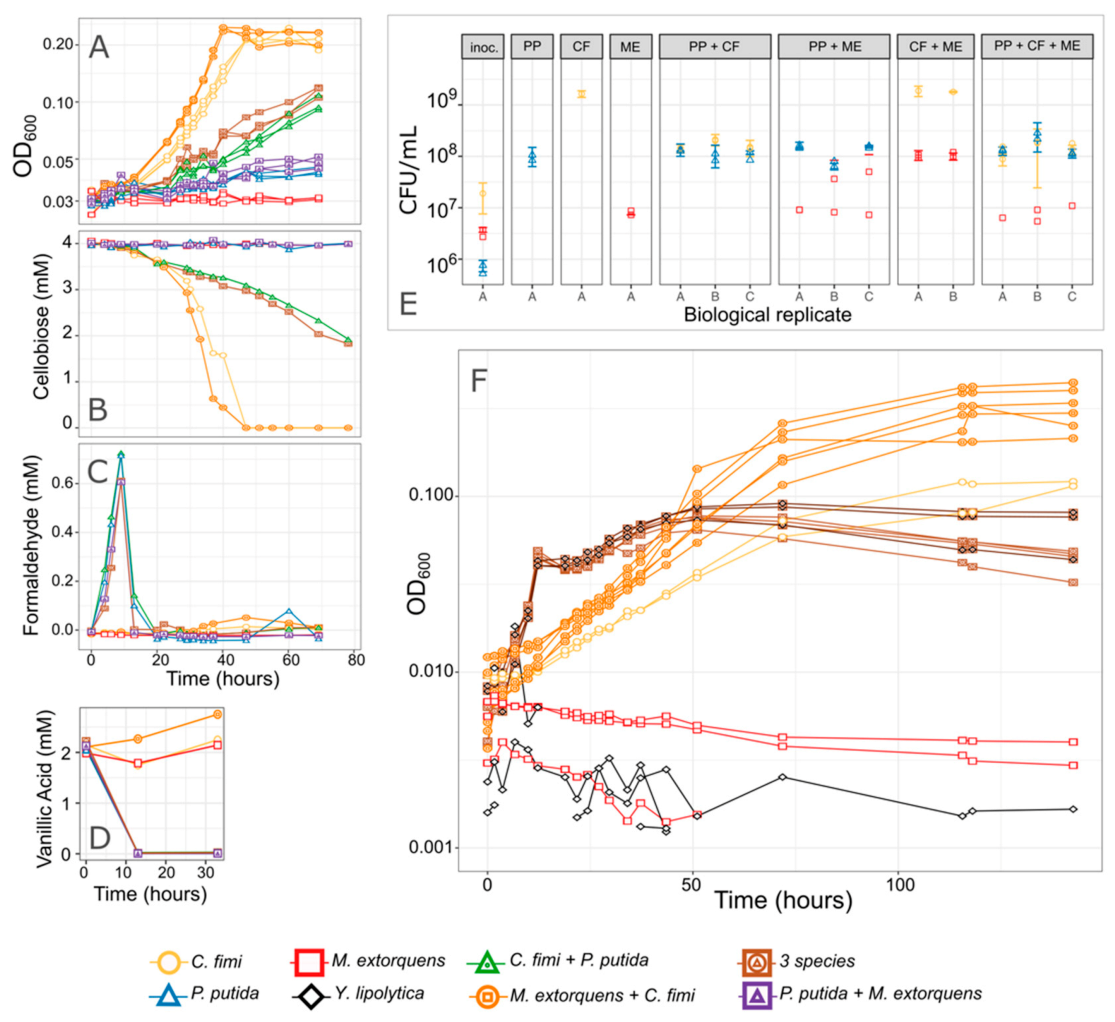
3.2. M. extorquens Reduces the Formaldehyde Concentrations in Cocultures Growing on Vanillic Acid
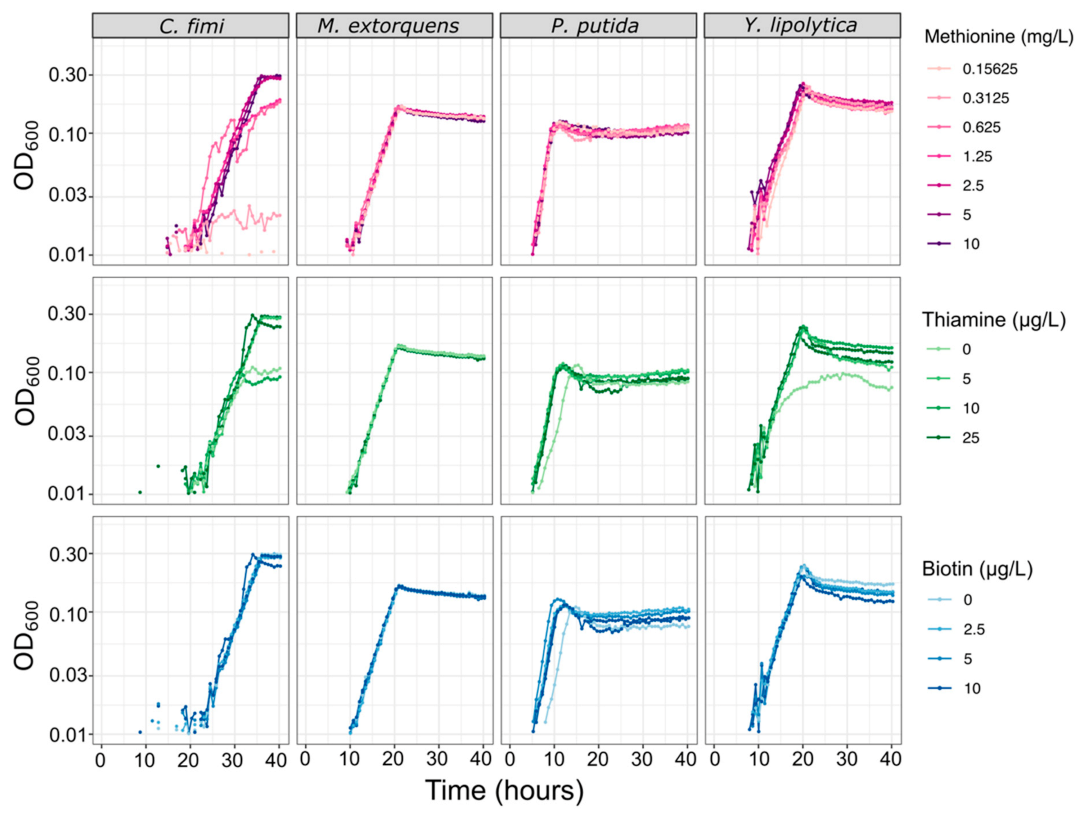
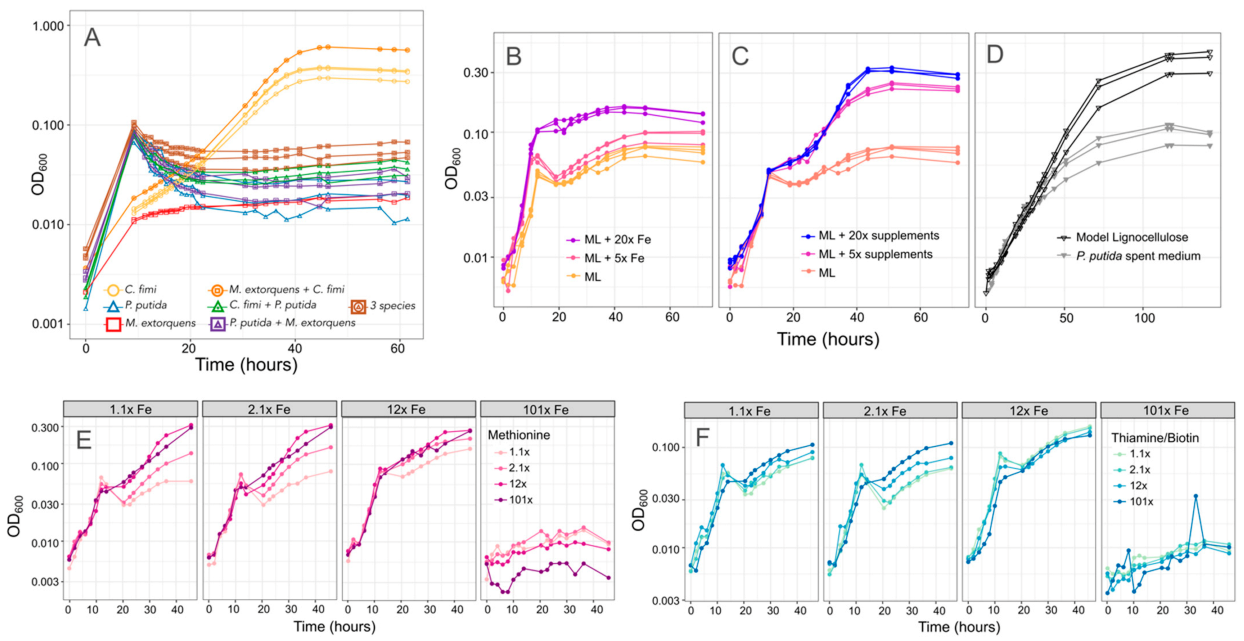
3.3. A Minimal Growth Medium Can Support All Members and Facilitates Metabolomic Analysis, with Modest Amino Acid and Vitamin Supplements and Reduced Buffer Concentrations
3.4. P. putida Inhibits Growth of C. fimi in Model Lignocellulose Medium, and C. fimi Supports the Growth of M. extorquens
3.5. P. putida Inhibition of C. fimi Growth May Be due to Multiple Mechanisms
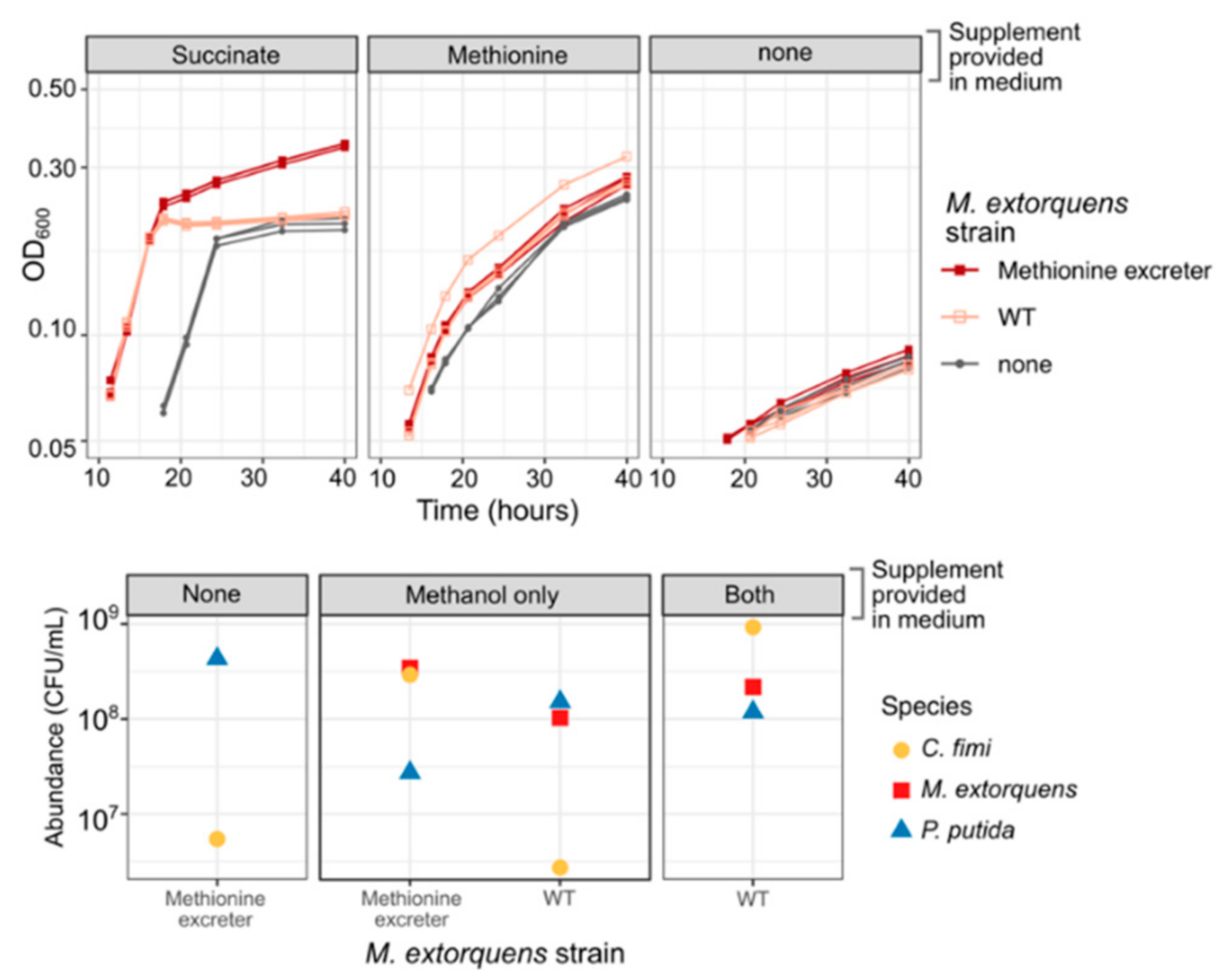
3.6. Methionine-Overproducing M. extorquens Can Support the Growth of C. fimi
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raud, M.; Kikas, T.; Sippula, O.; Shurpali, N.J. Potentials and challenges in lignocellulosic biofuel production technology. Renew. Sustain. Energy Rev. 2019, 111, 44–56. [Google Scholar] [CrossRef]
- Brodin, M.; Vallejos, M.; Opedal, M.T.; Area, M.C.; Chinga-Carrasco, G. Lignocellulosics as sustainable resources for production of bioplastics—A review. J. Clean. Prod. 2017, 162, 646–664. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Beckham, G.T.; Johnson, C.W.; Karp, E.M.; Salvachúa, D.; Vardon, D.R. Opportunities and challenges in biological lignin valorization. Curr. Opin. Biotechnol. 2016, 42, 40–53. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Brunel, F.; Davison, J. Cloning and sequencing of Pseudomonas genes encoding vanillate demethylase. J. Bacteriol. 1988, 170, 4924–4930. [Google Scholar] [CrossRef]
- Mitsui, R.; Kusano, Y.; Yurimoto, H.; Sakai, Y.; Kato, N.; Tanaka, M. Formaldehyde fixation contributes to detoxification for growth of a nonmethylotroph, Burkholderia cepacia TM1, on vanillic acid. Appl. Environ. Microbiol. 2003, 69, 6128–6132. [Google Scholar] [CrossRef]
- Sudtachat, N.; Ito, N.; Itakura, M.; Masuda, S.; Eda, S.; Mitsui, H.; Kawaharada, Y.; Minamisawa, K. Aerobic vanillate degradation and C1 compound metabolism in Bradyrhizobium japonicum. Appl. Environ. Microbiol. 2009, 75, 5012–5017. [Google Scholar] [CrossRef]
- Merkens, H.; Beckers, G.; Wirtz, A.; Burkovski, A. Vanillate metabolism in Corynebacterium glutamicum. Curr. Microbiol. 2005, 51, 59–65. [Google Scholar] [CrossRef]
- Chen, H.-P.; Chow, M.; Liu, C.-C.; Lau, A.; Liu, J.; Eltis, L.D. Vanillin catabolism in Rhodococcus jostii RHA1. Appl. Environ. Microbiol. 2012, 78, 586–588. [Google Scholar] [CrossRef]
- Daims, H.; Taylor, M.W.; Wagner, M. Wastewater treatment: A model system for microbial ecology. Trends Biotechnol. 2006, 24, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Bond-Watts, B.B.; Chang, M.C. Production of advanced biofuels in engineered E. coli. Curr. Opin. Chem. Biol. 2013, 17, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Zuroff, T.R.; Curtis, W.R. Developing symbiotic consortia for lignocellulosic biofuel production. Appl. Microbiol. Biotechnol. 2012, 93, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Marquez, J.C.F.; Do Nascimento, M.; Zehr, J.P.; Curatti, L. Genetic engineering of multispecies microbial cell factories as an alternative for bioenergy production. Trends Biotechnol. 2013, 31, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Minty, J.J.; Singer, M.E.; Scholz, S.A.; Bae, C.-H.; Ahn, J.-H.; Foster, C.E.; Liao, J.C.; Lin, X.N. Design and characterization of synthetic fungal-bacterial consortia for direct production of isobutanol from cellulosic biomass. Proc. Natl. Acad. Sci. USA 2013, 110, 14592–14597. [Google Scholar] [CrossRef]
- Khazaei, T.; Williams, R.L.; Bogatyrev, S.R.; Doyle, J.C.; Henry, C.S.; Ismagilov, R.F. Metabolic multistability and hysteresis in a model aerobe-anaerobe microbiome community. Sci. Adv. 2020, 6, eaba0353. [Google Scholar] [CrossRef]
- Cavka, A.; Stagge, S.; Jönsson, L.J. Identification of small aliphatic aldehydes in pretreated lignocellulosic feedstocks and evaluation of their inhibitory effects on yeast. J. Agric. Food Chem. 2015, 63, 9747–9754. [Google Scholar] [CrossRef]
- Brandt, B.A.; Jansen, T.; Görgens, J.F.; van Zyl, W.H. Overcoming lignocellulose-derived microbial inhibitors: Advancing the Saccharomyces cerevisiae resistance toolbox. Biofuels Bioprod. Biorefin. 2019, 13, 1520–1536. [Google Scholar] [CrossRef]
- Hibi, M.; Sonoki, T.; Mori, H. Functional coupling between vanillate-o-demethylase and formaldehyde detoxification pathway. FEMS Microbiol. Lett. 2005, 253, 237–242. [Google Scholar] [CrossRef]
- Nayak, D.D.; Marx, C.J. Genetic and phenotypic comparison of facultative methylotrophy between Methylobacterium extorquens Strains PA1 and AM1. PLoS ONE 2014, 9, e107887. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L.; Lidstrom, P.M.E. Aerobic methylotrophic prokaryotes. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 267–285. ISBN 978-3-642-30140-7. [Google Scholar]
- Lee, J.A.; Stolyar, S.; Marx, C.J. An aerobic link between lignin degradation and C1 metabolism: Growth on methoxylated aromatic compounds by members of the genus Methylobacterium. bioRxiv 2019, 712836. [Google Scholar] [CrossRef]
- Chubiz, L.M.; Purswani, J.; Carroll, S.; Marx, C.J. A novel pair of inducible expression vectors for use in Methylobacterium extorquens. BMC Res. Notes 2013, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Schneider, K.; Kiefer, P.; Massou, S.; Vorholt, J.A.; Portais, J.-C. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 2011, 5, 189. [Google Scholar] [CrossRef]
- Delaney, N.F.; Kaczmarek, M.E.; Ward, L.M.; Swanson, P.K.; Lee, M.-C.; Marx, C.J. Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS ONE 2013, 8, e62957. [Google Scholar] [CrossRef]
- Ravi, K.; García-Hidalgo, J.; Gorwa-Grauslund, M.F.; Lidén, G. Conversion of lignin model compounds by Pseudomonas putida KT2440 and isolates from compost. Appl. Microbiol. Biotechnol. 2017, 101, 5059–5070. [Google Scholar] [CrossRef]
- Belda, E.; van Heck, R.G.A.; José Lopez-Sanchez, M.; Cruveiller, S.; Barbe, V.; Fraser, C.; Klenk, H.-P.; Petersen, J.; Morgat, A.; Nikel, P.I.; et al. The revisited genome of Pseudomonas putida KT2440 enlightens its value as a robust metabolic chassis. Environ. Microbiol. 2016. [Google Scholar] [CrossRef]
- Simon, O.; Klaiber, I.; Huber, A.; Pfannstiel, J. Comprehensive proteome analysis of the response of Pseudomonas putida KT2440 to the flavor compound vanillin. J. Proteomics 2014, 109, 212–227. [Google Scholar] [CrossRef]
- Roca, A.; Rodríguez-Herva, J.J.; Ramos, J.L. Redundancy of enzymes for formaldehyde detoxification in Pseudomonas putida. J. Bacteriol. 2009, 191, 3367–3374. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Khawdas, W.; Aso, Y.; Ohara, H. Microbial fuel cells using Cellulomonas spp. with cellulose as fuel. J. Biosci. Bioeng. 2017, 123, 358–363. [Google Scholar] [CrossRef]
- Christopherson, M.R.; Suen, G.; Bramhacharya, S.; Jewell, K.A.; Aylward, F.O.; Mead, D.; Brumm, P.J. The genome sequences of Cellulomonas fimi and “Cellvibrio gilvus” reveal the cellulolytic strategies of two facultative anaerobes, transfer of “Cellvibrio gilvus” to the genus Cellulomonas, and proposal of Cellulomonas gilvus sp. nov. PLoS ONE 2013, 8, e53954. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, A.; Bowen, B.P.; Northen, T. Exometabolomic analysis of cross-feeding metabolites. Metabolites 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Blazeck, J.; Hill, A.; Liu, L.; Knight, R.; Miller, J.; Pan, A.; Otoupal, P.; Alper, H.S. Harnessing Yarrowia lipolytica lipogenesis to create a platform for lipid and biofuel production. Nat. Commun. 2014, 5, 3131. [Google Scholar] [CrossRef] [PubMed]
- Bredeweg, E.L.; Pomraning, K.R.; Dai, Z.; Nielsen, J.; Kerkhoven, E.J.; Baker, S.E. A Molecular genetic toolbox for Yarrowia lipolytica. Biotechnol. Biofuels 2017, 10, 2. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Segrè, D. Synthetic ecology of microbes: Mathematical models and applications. J. Mol. Biol. 2016, 428, 837–861. [Google Scholar] [CrossRef]
- Bazurto, J.; Nayak, D.D.; Ticak, T.; Davlieva, M.; Lee, J.A.; Lambert, L.B.; Benski, O.J.; Quates, C.J.; Johnson, J.L.; Patel, J.S.; et al. EfgA is a conserved formaldehyde sensor that halts bacterial translation in response to elevated formaldehyde. bioRxiv 2020. [Google Scholar] [CrossRef]
- Harcombe, W. Novel cooperation experimentally evolved between species. Evol. Int. J. Org. Evol. 2010, 64, 2166–2172. [Google Scholar] [CrossRef]
- Lawrence, D.A.; Smith, D.A.; Rowbury, R.J. Regulation of methionine synthesis in Salmonella typhimurium: Mutants resistant to inhibition by analogues of methionine. Genetics 1968, 58, 473–492. [Google Scholar] [CrossRef]
- Atlas, R.M. Handbook of Media for Environmental Microbiology, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-1-4200-3748-7. [Google Scholar]
- Nash, T. The colorimetric estimation of formaldehyde by means of the hantzsch reaction. Biochem. J. 1953, 55, 416–421. [Google Scholar] [CrossRef]
- Stalikas, C.D. Extraction, separation, and detection methods for phenolic acids and flavonoids. J. Sep. Sci. 2007, 30, 3268–3295. [Google Scholar] [CrossRef]
- Lee, J.A.; Riazi, S.; Nemati, S.; Bazurto, J.V.; Vasdekis, A.E.; Ridenhour, B.J.; Remien, C.H.; Marx, C.J. Microbial phenotypic heterogeneity in response to a metabolic toxin: Continuous, dynamically shifting distribution of formaldehyde tolerance in Methylobacterium extorquens populations. PLoS Genet. 2019, 15, e1008458. [Google Scholar] [CrossRef]
- Whittle, D.J.; Kilburn, D.G.; Warren, R.A.J.; Miller, R.C. Molecular cloning of a Cellulomonas fimi cellulase gene in Escherichia coli: Recombinant DNA; plasmid PBR322; immunoassay. Gene 1982, 17, 139–145. [Google Scholar] [CrossRef]
- Summers, R.J.; Boudreaux, D.P.; Srinivasan, V.R. Continuous cultivation for apparent optimization of defined media for Cellulomonas sp. and Bacillus cereus. Appl. Environ. Microbiol. 1979, 38, 66–71. [Google Scholar] [CrossRef]
- Gasmi, N.; Ayed, A.; Nicaud, J.-M.; Kallel, H. Design of an efficient medium for heterologous protein production in Yarrowia lipolytica: Case of human interferon alpha 2b. Microb. Cell Factories 2011, 10, 38. [Google Scholar] [CrossRef]
- Kebabci, Ö.; Cihangir, N. Comparison of three Yarrowia lipolytica strains for lipase production: NBRC 1658, IFO 1195, and a local strain. Turk. J. Biol. 2012, 36, 15–24. [Google Scholar]
- Han, J.S.; Rowell, J.S. Chemical composition of fibers, chapter 5. In Paper and Composites from Agro-Based Resources; Rowell, R.M., Young, R.A., Rowell, J.K., Eds.; CRC Press: Boca Raton, FL, USA, 1997; pp. 83–134. [Google Scholar]
- Buyer, J.S.; Sikora, L.J. Rhizosphere interactions and siderophores. Plant Soil 1990, 129, 101–107. [Google Scholar] [CrossRef]
- Joshi, H.; Dave, R.; Venugopalan, V.P. Pumping iron to keep fit: Modulation of siderophore secretion helps efficient aromatic utilization in Pseudomonas putida KT2440. Microbiology 2014, 160, 1393–1400. [Google Scholar] [CrossRef]
- Vorholt, J.A.; Chistoserdova, L.; Lidstrom, M.E.; Thauer, R.K. The NADP-dependent methylene tetrahydromethanopterin dehydrogenase in Methylobacterium extorquens AM1. J. Bacteriol. 1998, 180, 5351–5356. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.J.; Chistoserdova, L.; Lidstrom, M.E. Formaldehyde-detoxifying role of the tetrahydromethanopterin-linked pathway in Methylobacterium extorquens AM1. J. Bacteriol. 2003, 185, 7160–7168. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Riehl, W.J.; Dukovski, I.; Granger, B.R.; Betts, A.; Lang, A.H.; Bonilla, G.; Kar, A.; Leiby, N.; Mehta, P.; et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014, 7, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Granato, E.T.; Foster, K.R. The evolution of mass cell suicide in bacterial warfare. Curr. Biol. 2020, 30, 2836–2843.e3. [Google Scholar] [CrossRef]
- LaSarre, B.; McCully, A.L.; Lennon, J.T.; McKinlay, J.B. Microbial mutualism dynamics governed by dose-dependent toxicity of cross-fed nutrients. ISME J. 2017, 11, 337–348. [Google Scholar] [CrossRef]
- Gonzalez, D.; Sabnis, A.; Foster, K.R.; Mavridou, D.A.I. Costs and benefits of provocation in bacterial warfare. Proc. Natl. Acad. Sci. USA 2018, 115, 7593–7598. [Google Scholar] [CrossRef] [PubMed]
- Harcombe, W.R.; Chacón, J.M.; Adamowicz, E.M.; Chubiz, L.M.; Marx, C.J. Evolution of bidirectional costly mutualism from byproduct consumption. Proc. Natl. Acad. Sci. USA 2018, 115, 12000–12004. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Chubiz, L.M.; Harcombe, W.R.; Marx, C.J. Identification of the potentiating mutations and synergistic epistasis that enabled the evolution of inter-species cooperation. PLoS ONE 2017, 12, e0174345. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Component | Final Concentration |
|---|---|---|
| Basic modified PIPES medium (MP) | PIPES free acid | 3 mM [30 mM] |
| Potassium phosphate dibasic, K2HPO4 | 2.90 mM [1.45 mM] | |
| Sodium phosphate monobasic, NaH2PO4 | 3. 76 mM [1.88 mM] | |
| Magnesium chloride, MgCl2 | 0.5 mM | |
| Ammonium sulfate, (NH4)2SO4 | 5 mM | |
| Sodium citrate, Na3C6H5O7 | 45.286 µM | |
| Zinc sulfate, ZnSO4 | 1.2 µM | |
| Manganese chloride, MnCl2 | 1.02 µM | |
| Iron(II) sulfate, FeSO4 | 17.768 µM | |
| Ammonium heptamolybdate, (NH4)6Mo7O24 | 2 µM | |
| Copper(II) sulfate, CuSO4 | 1 µM | |
| Cobalt chloride, CoCl2 | 2 µM | |
| Sodium tungstate, Na2WO4 | 0.338 µM | |
| Calcium chloride, CaCl2 | 0.02 mM | |
| Supplements | Methionine | 2 mg/L |
| Thiamine | 5 µg/L | |
| Biotin | 40 µg/L | |
| Carbon substrates | Cellobiose | 4 mM |
| Xylose | 5 mM | |
| Vanillic Acid | 4 mM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.A.; Baugh, A.C.; Shevalier, N.J.; Strand, B.; Stolyar, S.; Marx, C.J. Cross-Feeding of a Toxic Metabolite in a Synthetic Lignocellulose-Degrading Microbial Community. Microorganisms 2021, 9, 321. https://doi.org/10.3390/microorganisms9020321
Lee JA, Baugh AC, Shevalier NJ, Strand B, Stolyar S, Marx CJ. Cross-Feeding of a Toxic Metabolite in a Synthetic Lignocellulose-Degrading Microbial Community. Microorganisms. 2021; 9(2):321. https://doi.org/10.3390/microorganisms9020321
Chicago/Turabian StyleLee, Jessica A., Alyssa C. Baugh, Nicholas J. Shevalier, Brandi Strand, Sergey Stolyar, and Christopher J. Marx. 2021. "Cross-Feeding of a Toxic Metabolite in a Synthetic Lignocellulose-Degrading Microbial Community" Microorganisms 9, no. 2: 321. https://doi.org/10.3390/microorganisms9020321
APA StyleLee, J. A., Baugh, A. C., Shevalier, N. J., Strand, B., Stolyar, S., & Marx, C. J. (2021). Cross-Feeding of a Toxic Metabolite in a Synthetic Lignocellulose-Degrading Microbial Community. Microorganisms, 9(2), 321. https://doi.org/10.3390/microorganisms9020321