
DNA Methylation Patterns Differ between Free-Living Rhizobium leguminosarum RCAM1026 and Bacteroids Formed in Symbiosis with Pea (Pisum sativum L.)

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
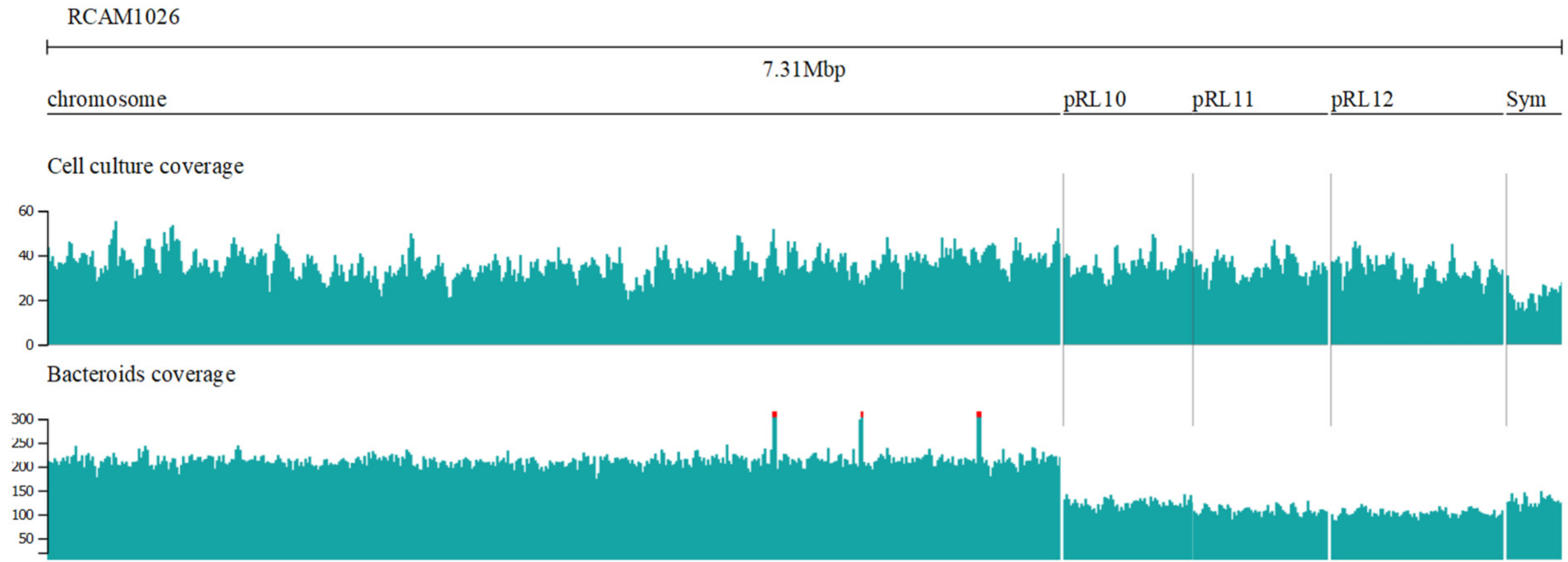
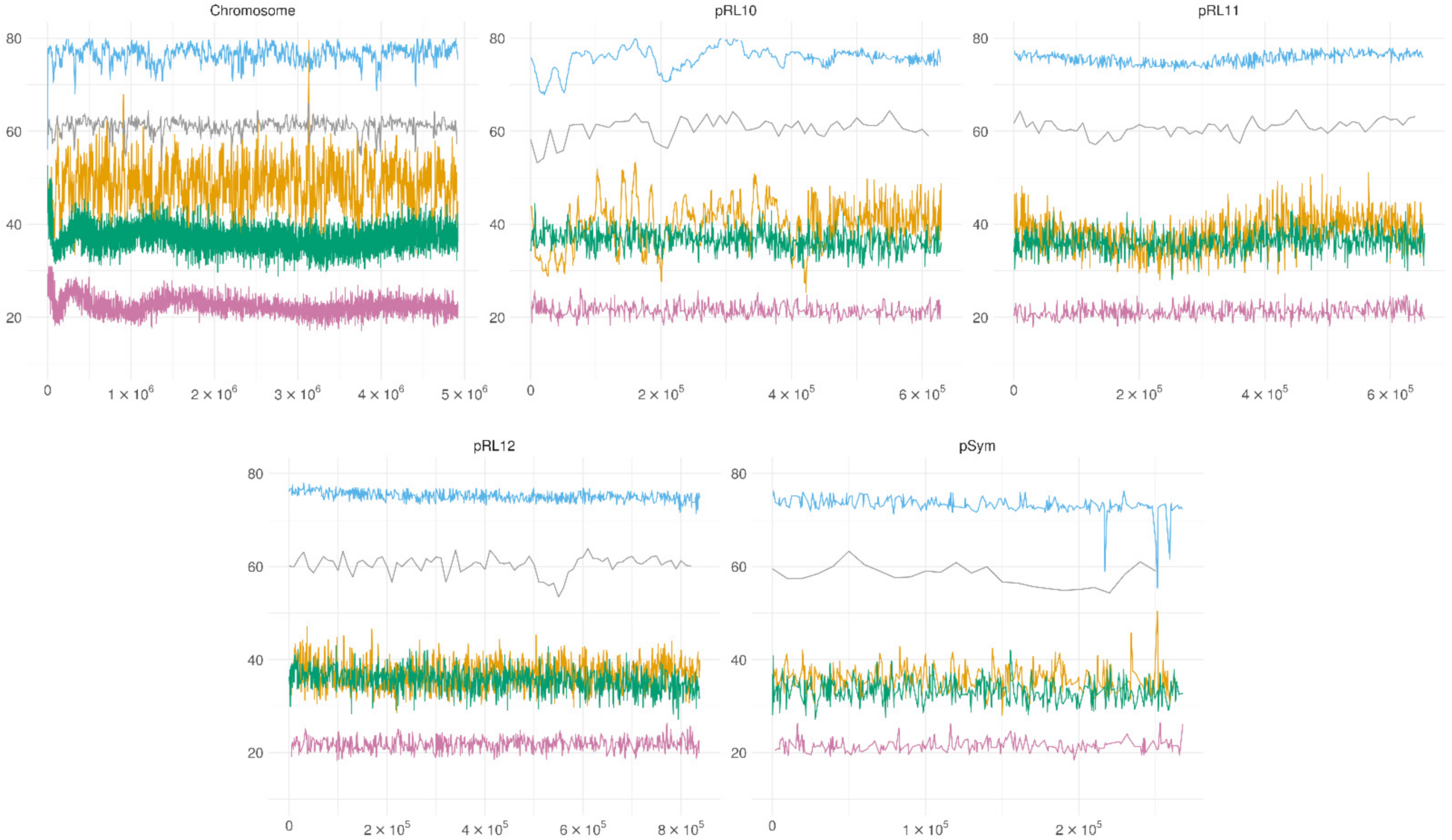
3.1. Genome Assembly and Comparison
3.2. Methylation Motif Search
3.3. Methylation System Genes in the RCAM1026 Genome
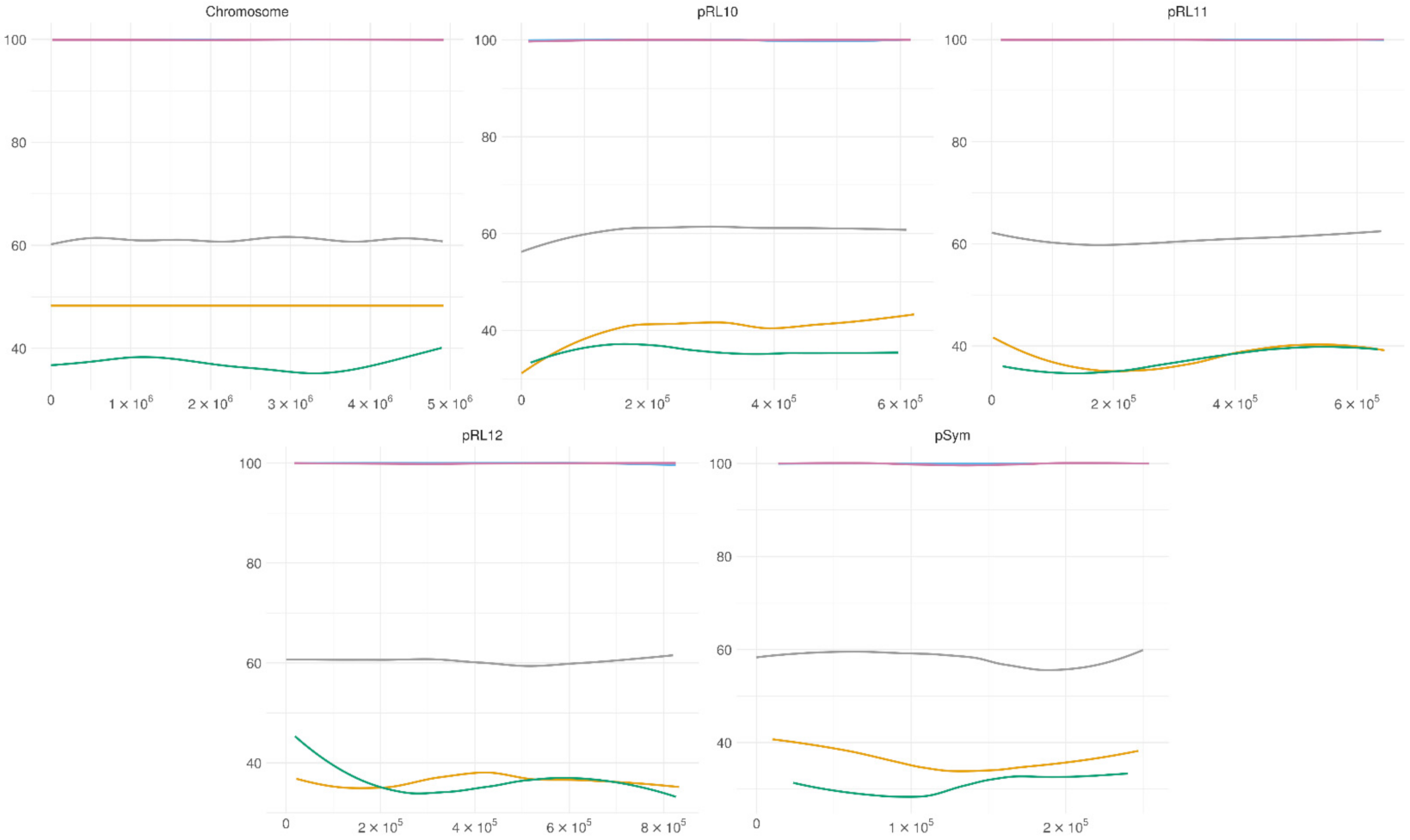
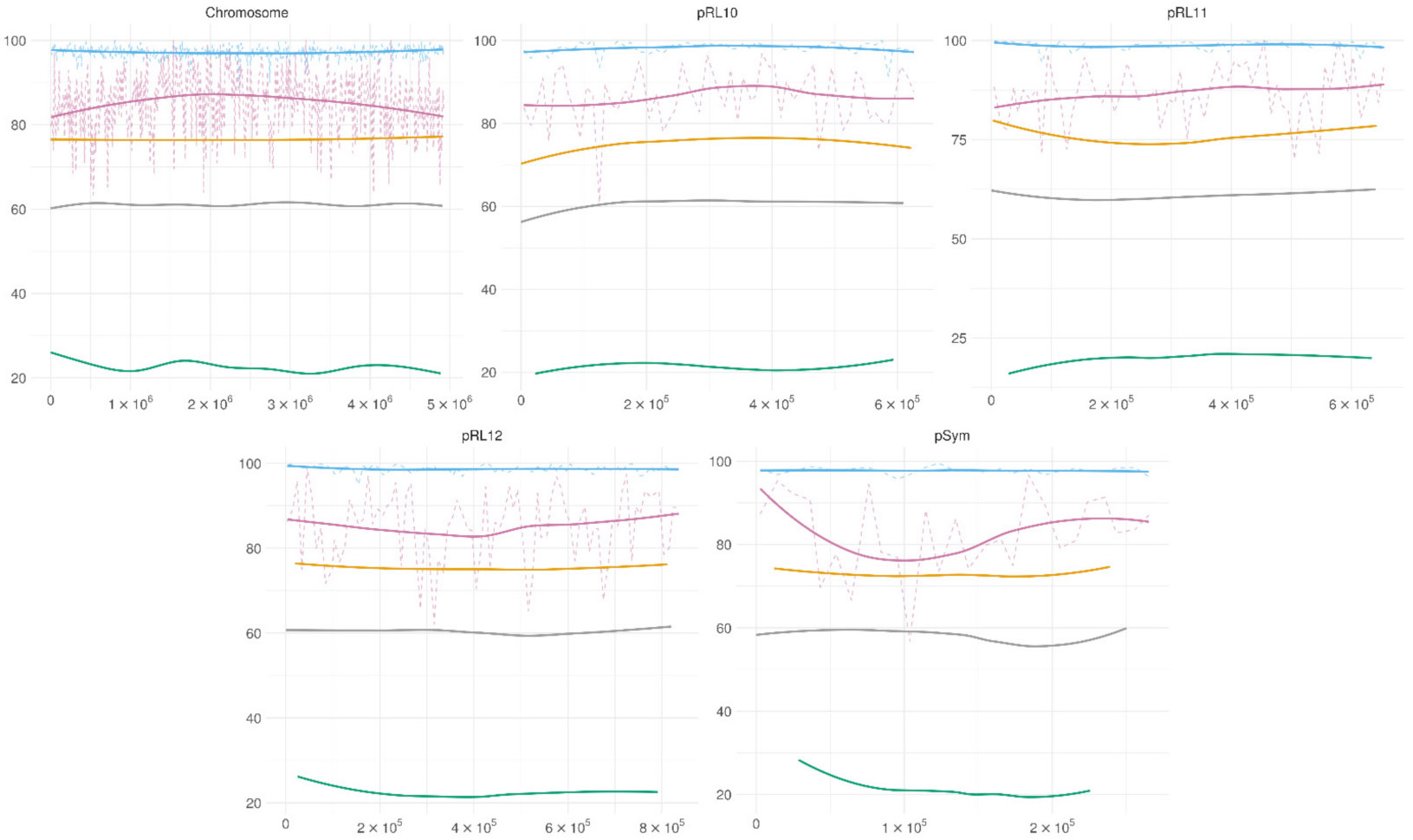
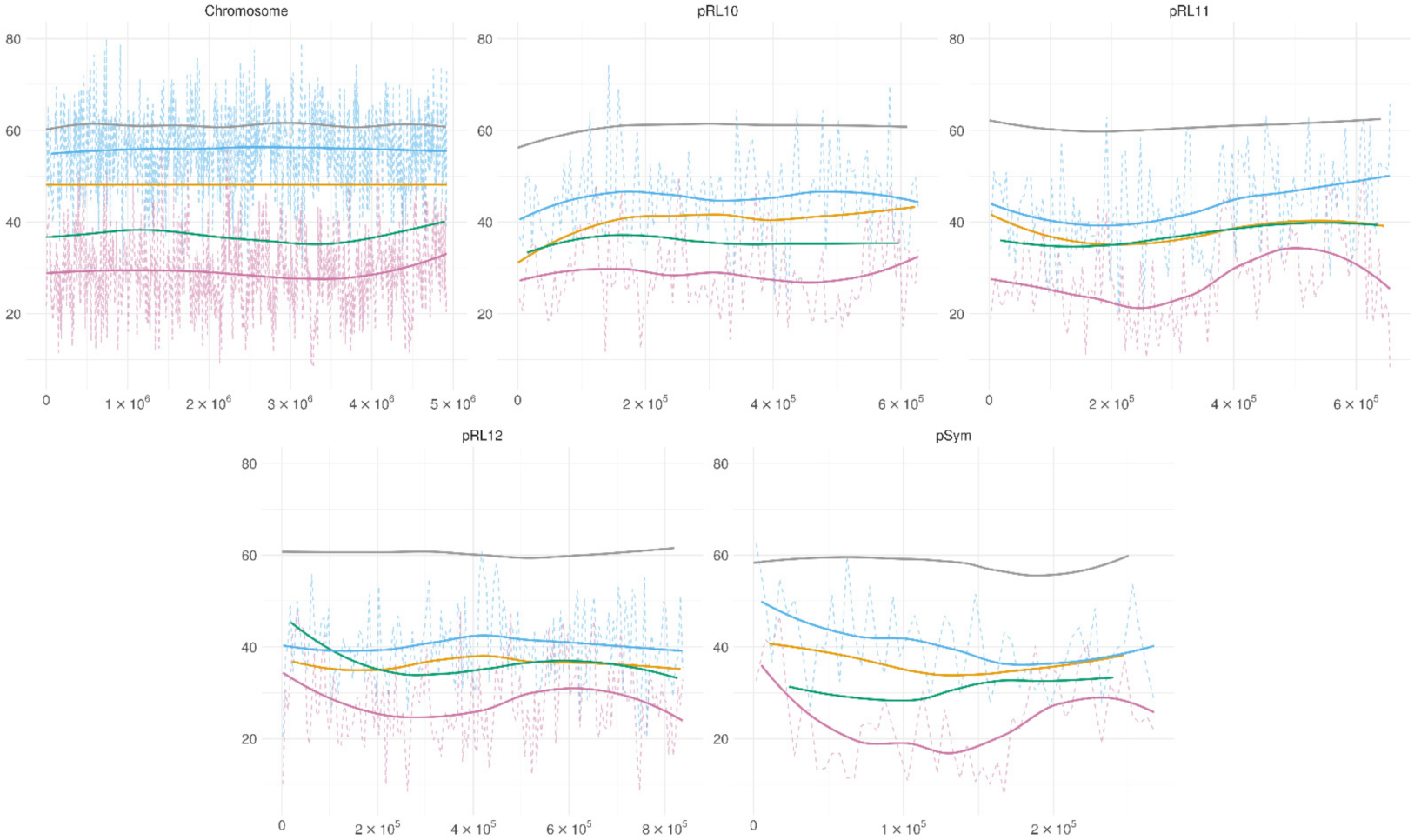
3.4. Genome-Wide Methylation Patterns
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bickle, T.A.; Krüger, D.H. Biology of DNA restriction. Microbiol. Rev. 1993, 57, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Collier, J. Epigenetic regulation of the bacterial cell cycle. Curr. Opin. Microbiol. 2009, 12, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Beaulaurier, J.; Schadt, E.E.; Fang, G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet. 2019, 20, 157–172. [Google Scholar] [CrossRef]
- Casadesús, J.; Low, D. Epigenetic Gene Regulation in the Bacterial World. Microbiol. Mol. Biol. Rev. 2006, 70, 830–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boye, E.; Løbner-Olesen, A.; Skarstad, K. Limiting DNA replication to once and only once. EMBO Rep. 2000, 1, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Løbner-Olesen, A.; Marinus, M.G.; Hansen, F.G. Role of SeqA and Dam in Escherichia coli gene expression: A global/microarray analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 4672–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, D.A.; Casadesús, J. Clocks and switches: Bacterial gene regulation by DNA adenine methylation. Curr. Opin. Microbiol. 2008, 11, 106–112. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Alexander, N.; Grigorev, K.; Bezdan, D.; Sichtig, H.; Chiu, C.Y.; Mason, C.E. Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat. Commun. 2019, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Spadar, A.; Perdigão, J.; Phelan, J.; Charleston, J.; Modesto, A.; Elias, R.; de Sessions, P.F.; Hibberd, M.L.; Campino, S.; Duarte, A.; et al. Methylation analysis of Klebsiella pneumoniae from Portuguese hospitals. Sci. Rep. 2021, 11, 6491. [Google Scholar] [CrossRef]
- Downie, J.A. Legume nodulation. Curr. Biol. 2014, 24, R184–R190. [Google Scholar] [CrossRef] [Green Version]
- De La Peña, T.C.; Fedorova, E.; Pueyo, J.J.; Lucas, M.M. The Symbiosome: Legume and Rhizobia Co-evolution toward a Nitrogen-Fixing Organelle? Front. Plant Sci. 2018, 8, 2229. [Google Scholar] [CrossRef] [Green Version]
- Remigi, P.; Zhu, J.; Young, J.P.W.; Masson-Boivin, C. Symbiosis within Symbiosis: Evolving Nitrogen-Fixing Legume Symbionts. Trends Microbiol. 2016, 24, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Sprent, J.; Ardley, J.; James, E. From North to South: A latitudinal look at legume nodulation processes. S. Afr. J. Bot. 2013, 89, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Alunni, B.; Gourion, B. Terminal bacteroid differentiation in the legume−rhizobium symbiosis: Nodule-specific cysteine-rich peptides and beyond. New Phytol. 2016, 211, 411–417. [Google Scholar] [CrossRef] [Green Version]
- Ichida, H.; Matsuyama, T.; Abe, T.; Koba, T. DNA adenine methylation changes dramatically during establishment of symbiosis. FEBS J. 2007, 274, 951–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis-Richardson, A.G.; Russell, J.T.; Dias, R.; McKinlay, A.J.; Canepa, R.; Fagen, J.R.; Rusoff, K.T.; Drew, J.C.; Kolaczkowski, B.; Emerich, D.W.; et al. Corrigendum: Integrating DNA Methylation and Gene Expression Data in the Development of the Soybean-Bradyrhizobium N2-Fixing Symbiosis. Front. Microbiol. 2016, 7, 952. [Google Scholar] [CrossRef] [Green Version]
- Dicenzo, G.; Cangioli, L.; Nicoud, Q.; Cheng, J.H.T.; Blow, M.; Shapiro, N.; Woyke, T.; Biondi, E.; Alunni, B.; Mengoni, A.; et al. DNA Methylation in Ensifer Species during Free-Living Growth and during Nitrogen-Fixing Symbiosis with Medicago spp. bioRxiv 2021. [Google Scholar] [CrossRef]
- Afonin, A.; Sulima, A.; Zhernakov, A.; Zhukov, V. Draft genome of the strain RCAM1026 Rhizobium leguminosarum bv. viciae. Genom. Data 2017, 11, 85–86. [Google Scholar] [CrossRef]
- Afonin, A.M.; Leppyanen, I.V.; Kulaeva, O.A.; Shtark, O.Y.; Tikhonovich, I.A.; Dolgikh, E.A.; Zhukov, V.A. A high coverage reference transcriptome assembly of pea (Pisum sativum L.) mycorrhizal roots. Vavilov J. Genet. Breed. 2020, 24, 331–339. [Google Scholar] [CrossRef]
- Catalano, C.M.; Lane, W.S.; Sherrier, D.J. Biochemical characterization of symbiosome membrane proteins fromMedicago truncatula root nodules. Electrophorrsis 2004, 25, 519–531. [Google Scholar] [CrossRef]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Short Protocols in Molecular Biology: A Compendium of Methods from Current Protocols in Molecular Biology, 5th ed.; John Wiley & Sons: New York, NY, USA, 2002; ISBN 0471250929. [Google Scholar]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.M.; Gribchenko, E.S.; Sulima, A.S.; Zhukov, V.A. Complete Genome Sequence of an Efficient Rhizobium leguminosarum bv. viciae Strain, A1. Microbiol. Resour. Announc. 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Delcher, A.L.; Phillippy, A.; Coston, R.; Salzberg, S.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; Von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Liu, Y.; Jiang, Y.; Li, J.; Gao, Y.; Cui, Z.; Liu, Y.; Liu, B.; Wang, Y. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 2020, 21, 1–24. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tourancheau, A.; Mead, E.A.; Zhang, X.-S.; Fang, G. Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat. Methods 2021, 18, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Kristensen, D.M.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Microbial Genome Analysis: The COG Approach. Brief. Bioinform. 2019, 20, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- REBASE. The Restriction Enzyme Database. Available online: http://rebase.neb.com/rebase/rebase.htm (accessed on 30 August 2021).
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A Database for DNA Restriction and Modification: Enzymes, Genes and Genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef]
- Lapidus, A.; Korobeynikov, A. Selected Abstracts of “Bioinformatics: From Algorithms to Applications 2021” Conference. BMC Bioinform. 2021, 21, 1–18. [Google Scholar] [CrossRef]
- Heuer, H.; Abdo, Z.; Smalla, K. Patchy distribution of flexible genetic elements in bacterial populations mediates robustness to environmental uncertainty. FEMS Microbiol. Ecol. 2008, 65, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Wion, D.; Casadesús, J. N6-methyl-adenine: An epigenetic signal for DNA–protein interactions. Nat. Rev. Genet. 2006, 4, 183–192. [Google Scholar] [CrossRef]
- Ibryashkina, E.M.; Zakharova, M.V.; Baskunov, V.B.; Bogdanova, E.S.; Nagornykh, M.O.; Den’Mukhamedov, M.M.; Melnik, B.S.; Kolinski, A.; Gront, D.; Feder, M.; et al. Type II restriction endonuclease R.Eco29kI is a member of the GIY-YIG nuclease superfamily. BMC Struct. Biol. 2007, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Naito, T.; Kusano, K.; Kobayashi, I. Selfish Behavior of Restriction-Modification Systems. Science 1995, 267, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome Name | Length | CDS | tRNA Genes | GC Composition | Coverage in Bacteroids | Coverage in Cell Culture 1 |
|---|---|---|---|---|---|---|
| Chromosome | 4,921,456 | 4736 | 51 | 61.09 | 1 | 1 |
| pRL10 | 629,474 | 575 | 2 | 60.61 | 0.91 | 0.58 |
| pRL11 | 655,637 | 625 | - | 60.89 | 0.94 | 0.5 |
| pRL12 | 838,366 | 756 | - | 60.40 | 0.97 | 0.48 |
| Symbiotic plasmid | 268,924 | 263 | - | 58.12 | 0.59 | 0.6 |
| Motif | Characterized Motif | Modified Base | Modified Position | Nanodisco Score |
|---|---|---|---|---|
| GANTC | G6mANTC | 6mA | 2 | 34.16 |
| GATC | GAT4mC | 4mC | 4 | 35.68 |
| GGCGCC | GG4mCGCC | 4mC | 3 | 41.49 |
| Motif | Chromosome | pRL10 | pRL11 | pRL12 | pSym |
|---|---|---|---|---|---|
| GANTC | 6873 (841) 1 | 770 (96) | 778 (136) | 1037 (149) | 406 (65) |
| GATC | 45,690 (6029) | 5963 (633) | 6042 (595) | 7889 (767) | 2264 (252) |
| GGCGCC | 6466 (812) | 756 (45) | 754 (57) | 888 (81) | 213 (23) |
| Gene | Replicon | Putative Enzyme Type | Motif | Homologue in REBASE | Similarity |
|---|---|---|---|---|---|
| 000897 | Chromosome | Methyltransferase | - | M.MspCH12ORF7910P | 54.422 |
| 000974 | Chromosome | Methyltransferase | - | M.Hhe1ORF5290P | 59.823 |
| 000982 | Chromosome | Methyltransferase | GATC | M.MspME121ORFAP | 51.515 |
| 001161 | Chromosome | Methyltransferase | GANTC | M.RleNORF744P | 100.000 |
| 001164 | Chromosome | Nicking endonuclease | - | N.Pec32ORF2247P | 56.267 |
| 001182 | Chromosome | Methyltransferase | GGCGCC | M.CspK31ORF2261P | 70.984 |
| 001183 | Chromosome | Restriction enzyme | - | Avi39ORF4780P | 64.912 |
| 001184 | Chromosome | Restriction enzyme | - | Sma240ORF2946P | 75.431 |
| 001185 | Chromosome | Nicking endonuclease | - | V.OspA1ORF4070P | 69.919 |
| 001185 | Chromosome | Helicase domain protein | - | H.AspSLV7ORF8235P | 82.676 |
| 001515 | Chromosome | Methyltransferase | - | M.CspCJ34ORFGP | 50.882 |
| 001573 | Chromosome | Methyltransferase | - | M.Hhe1ORF5290P | 58.850 |
| 002613 | Chromosome | Orphan methyltransferase | GATC | M.Sen6759Dam | 50.935 |
| 002688 | Chromosome | Methyltransferase | - | M.EcoF3113ORF24645P | 57.277 |
| 003957 | Chromosome | Methyltransferase | - | M.EcoF3113ORF24645P | 68.584 |
| 005751 | pRl11 | Restriction enzyme | GGCGCC | M.SfrNXT3ORF1642P | 89.922 |
| 005752 | pRl11 | Methyltransferase | GGCGCC | SfrNXT3ORF1642P | 92.014 |
| 005755 | pRl11 | Nicking endonuclease | - | V.OspA1ORF4070P | 67.424 |
| 006861 | pRl12 | Methyltransferase | - | M.Hhe1ORF5290P | 60.526 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonin, A.M.; Gribchenko, E.S.; Zorin, E.A.; Sulima, A.S.; Zhukov, V.A. DNA Methylation Patterns Differ between Free-Living Rhizobium leguminosarum RCAM1026 and Bacteroids Formed in Symbiosis with Pea (Pisum sativum L.). Microorganisms 2021, 9, 2458. https://doi.org/10.3390/microorganisms9122458
Afonin AM, Gribchenko ES, Zorin EA, Sulima AS, Zhukov VA. DNA Methylation Patterns Differ between Free-Living Rhizobium leguminosarum RCAM1026 and Bacteroids Formed in Symbiosis with Pea (Pisum sativum L.). Microorganisms. 2021; 9(12):2458. https://doi.org/10.3390/microorganisms9122458
Chicago/Turabian StyleAfonin, Alexey M., Emma S. Gribchenko, Evgeny A. Zorin, Anton S. Sulima, and Vladimir A. Zhukov. 2021. "DNA Methylation Patterns Differ between Free-Living Rhizobium leguminosarum RCAM1026 and Bacteroids Formed in Symbiosis with Pea (Pisum sativum L.)" Microorganisms 9, no. 12: 2458. https://doi.org/10.3390/microorganisms9122458
APA StyleAfonin, A. M., Gribchenko, E. S., Zorin, E. A., Sulima, A. S., & Zhukov, V. A. (2021). DNA Methylation Patterns Differ between Free-Living Rhizobium leguminosarum RCAM1026 and Bacteroids Formed in Symbiosis with Pea (Pisum sativum L.). Microorganisms, 9(12), 2458. https://doi.org/10.3390/microorganisms9122458