
Modulation of the Tomato Rhizosphere Microbiome via Changes in Root Exudation Mediated by the Ethylene Receptor NR


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth
2.2. Experimental Soils
2.3. Collection of Root Exudates
2.4. LC-MS/MS Analysis, Data Preprocessing and Annotation of Root Exudates
2.5. Continuous and Nondestructive Sampling for Rhizosphere Soils of Tomato Plants
2.6. DNA Extraction and Metagenome Sequencing
2.7. Statistical Analyses
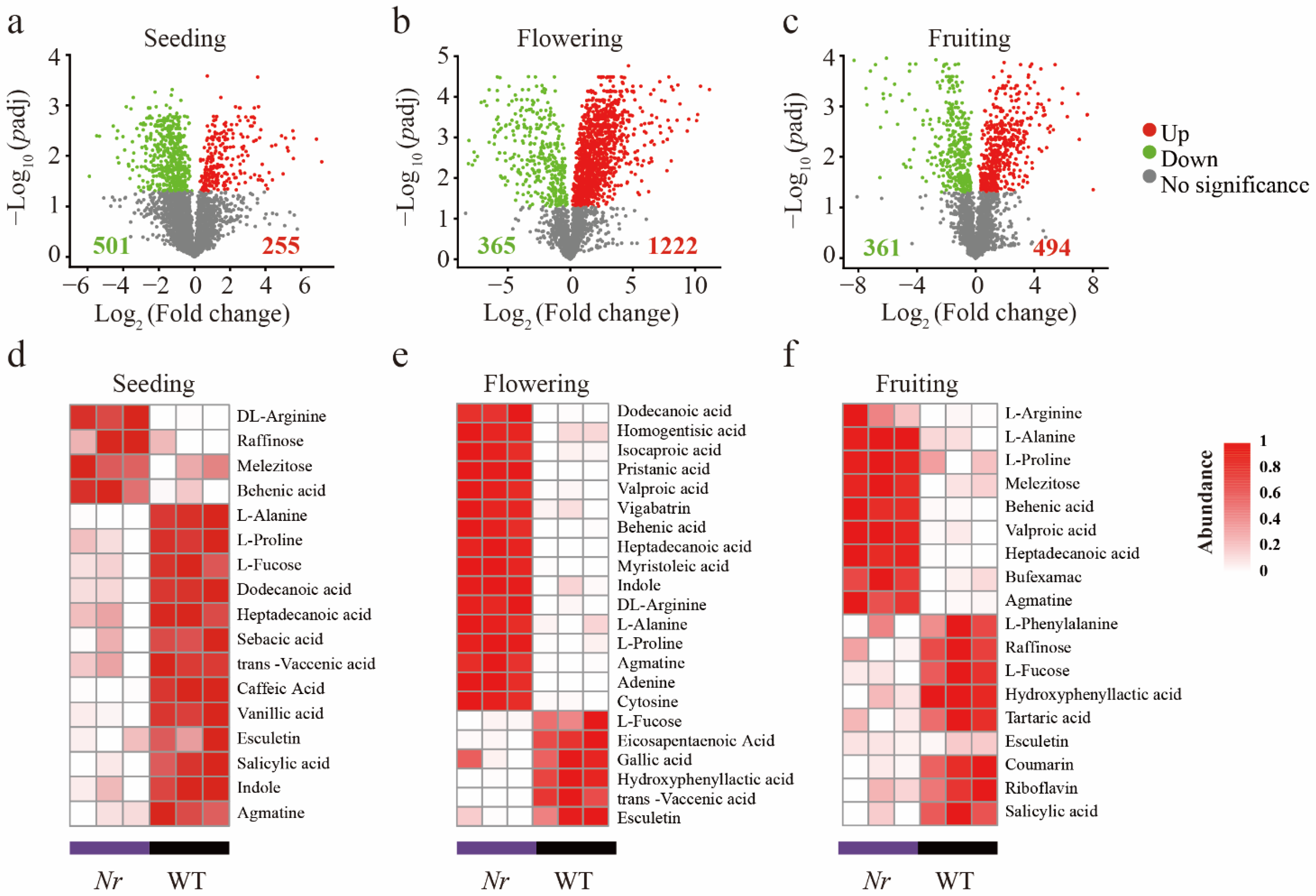
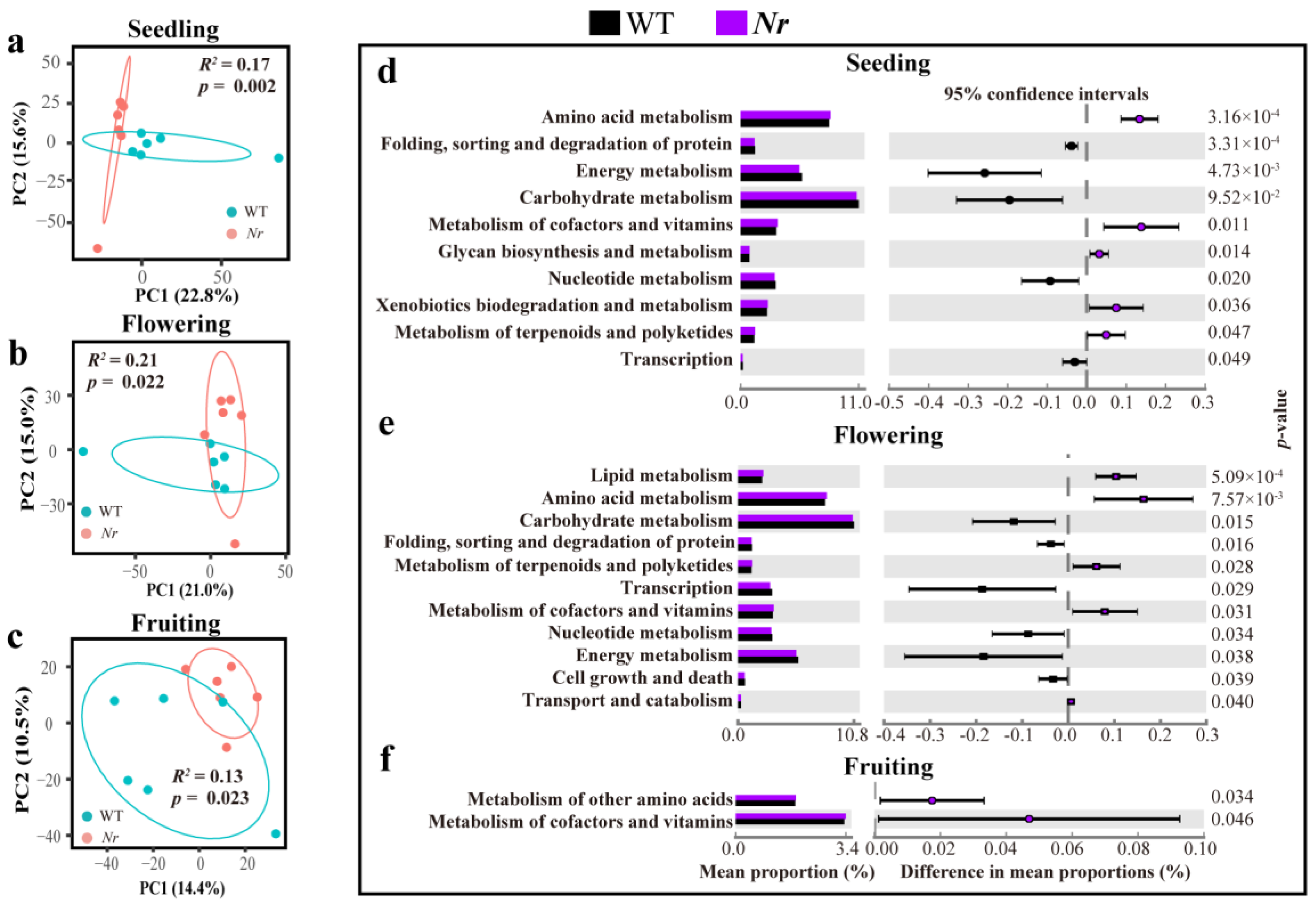
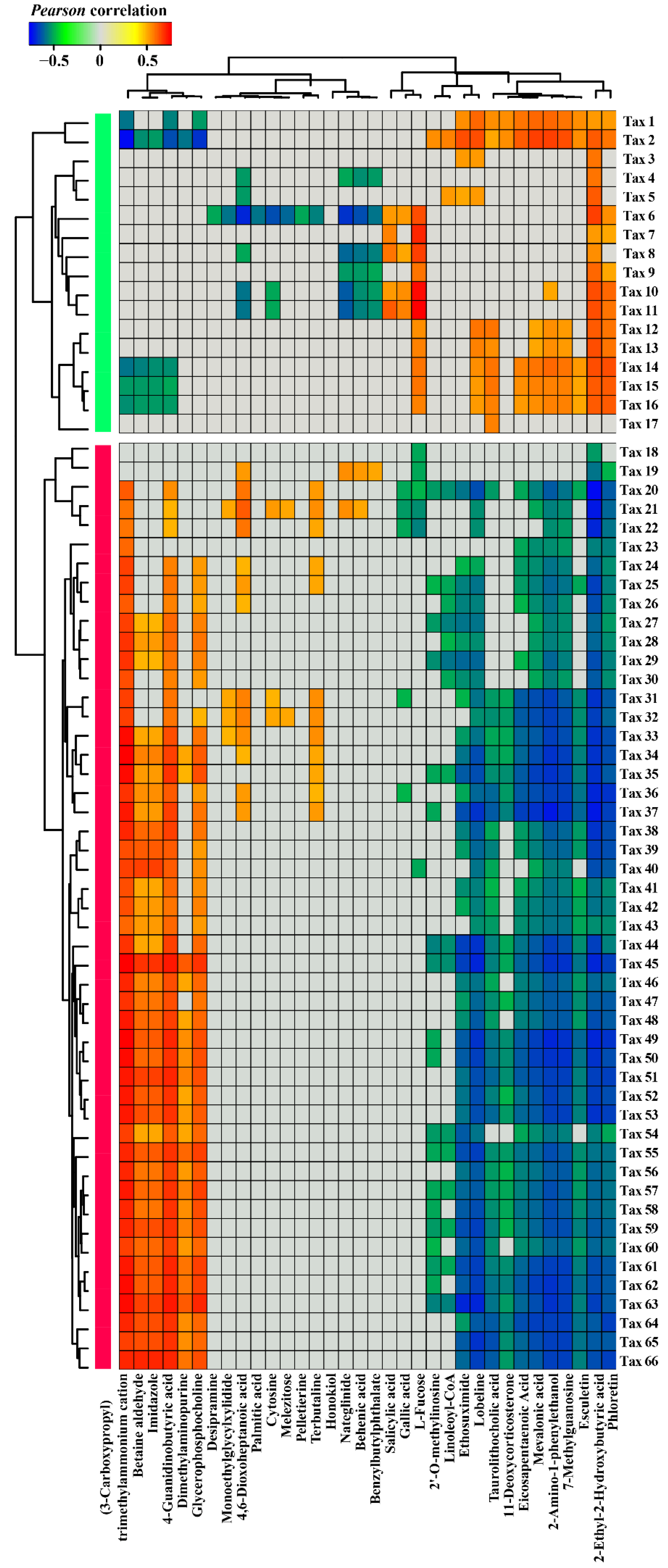
3. Results
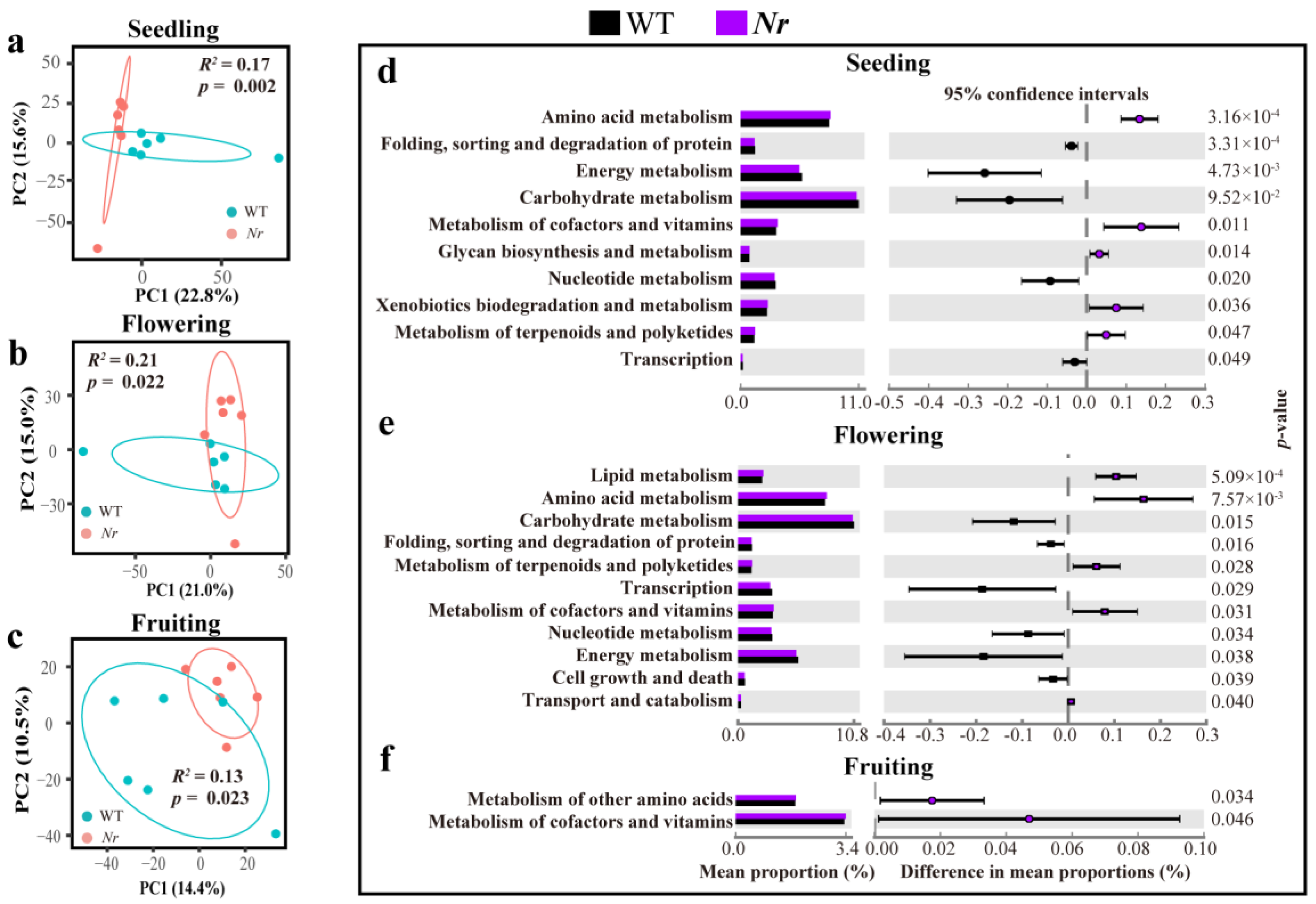
3.1. Chemical Characterization of WT and Nr Plants Exudates at Distinct Stages of Plant Development
3.2. Profiling the Root Microbiome of WT and Nr at Distinct Stages of Plant Development
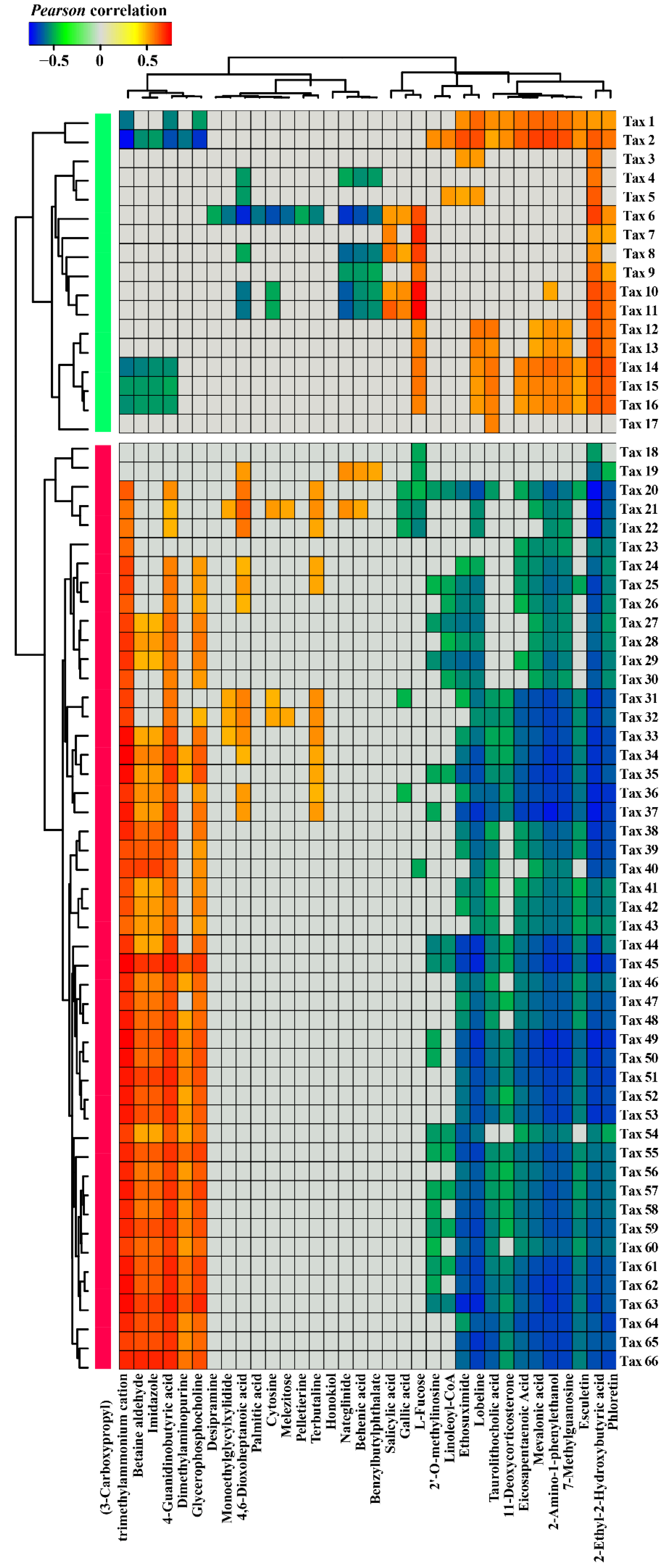
3.3. Correlations between Nr-related Bacterial Taxa and Root Exudate Metabolites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Pii, Y.; Mimmo, T.; Tomasi, N.; Terzano, R.; Cesco, S.; Crecchio, C. Microbial interactions in the rhizosphere: Beneficial influences of plant growth-promoting rhizobacteria on nutrient acquisition process. A review. Biol. Fertil. Soils 2015, 51, 403–415. [Google Scholar] [CrossRef]
- de Zelicourt, A.; AI-Yousif, M.; Hirt, H. Rhizosphere microbes as essential partners for plant stress tolerance. Mol. Plant 2013, 6, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Pineda, A.; Soler, R.; Pozo, M.J.; Rasmann, S.; Turlings, T.C.J. Above-belowground interactions involving plants, microbes and insects. Front. Plant Sci. 2015, 6, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente Cantó, C.; Simonin, M.; King, E.; Moulin, L.; Bennett, M.J.; Castrillo, G.; Laplaze, L. An extended root phenotype: The rhizosphere, its formation and impacts on plant fitness. Plant J. 2020, 103, 951–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.-X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Fitzpatrick, C.R.; Copeland, J.; Wang, P.W.; Guttman, D.S.; Kotanen, P.M.; Johnson, M.T.J. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. USA 2018, 115, 1157–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, J.; Gerin, F.; Le Gouis, J.; Moënne-Loccoz, Y.; Prigent–Combaret, C. Ancient wheat varieties have a higher ability to interact with plant growth-promoting rhizobacteria. Plant Cell Environ. 2020, 43, 246–260. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, M.; Ohno, K.; Takamatsu, K.; Aoki, Y.; Yamazaki, S.; Takase, H.; Shoji, T.; Yazaki, K.; Sugiyama, A. Tomato roots secrete tomatine to modulate the bacterial assemblage of the rhizosphere. Plant Physiol. 2021, 186, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Cotton, T.E.A.; Pétriacq, P.; Cameron, D.D.; Al Meselmani, M.; Schwarzenbacher, R.; Rolfe, S.A.; Ton, J. Metabolic regulation of the maize rhizobiome by benzoxazinoids. ISME J. 2019, 13, 1647–1658. [Google Scholar] [CrossRef] [Green Version]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; Da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, J.F., Jr.; McNickle, G.G.; Haag, J.J.; Lamb, E.G.; Nyanumba, S.M.; St Clair, C.C. Plants integrate information about nutrients and neighbors. Science 2010, 328, 1657. [Google Scholar] [CrossRef] [Green Version]
- Carvalhais, L.C.; Dennis, P.G.; Badri, D.V.; Kidd, B.N.; Vivanco, J.M.; Schenk, P.M. Linking jasmonic acid signaling, root exudates, and rhizosphere microbiomes. Mol. Plant-Microbe Interact. 2015, 28, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Bonkowski, M.; Shen, Y.; Griffiths, B.S.; Jiang, Y.; Wang, X.; Sun, B. Root ethylene mediates rhizosphere microbial community reconstruction when chemically detecting cyanide produced by neighbouring plants. Microbiome 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.X.; Ahammed, G.; Wu, C.; Fan, S.; Zhou, Y.-H. Crosstalk among jasmonate, salicylate and ethylene signaling pathways in plant disease and immune responses. Curr. Protein Pept. Sci. 2015, 16, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.C.; Li, H.; Ecker, J.R. Ethylene biosynthesis and signaling networks. Plant Cell 2002, 14, 131–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, C.; Shemansky, J.; Lin, D.Y.; Ying, Z.; Chang, J.; Garrett, W.M.; Kessenbrock, M.; Groth, G.; Tucker, M.; Cooper, B.; et al. CTR1 phosphorylates the central regulator EIN2 to control ethylene hormone signaling from the ER membrane to the nucleus in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 19486–19491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, P.R.; Ecker, J.R. The ethylene gas signal transduction pathway: A molecular perspective. Annu. Rev. Genet. 1998, 32, 227–254. [Google Scholar] [CrossRef] [PubMed]
- Benavente, L.M.; Alonso, J.M. Molecular mechanisms of ethylene signaling in Arabidopsis. Mol. Biosyst. 2006, 2, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Limagomes, B.; Mila, I.; Purgatto, E.; Peres, L.E.P.; Frasse, P.; Maza, E.; Zouine, M.; Roustan, J.P.; Bouzayen, M. Comprehensive profiling of ethylene response factor expression identifies ripening-associated ERF genes and their link to key regulators of fruit ripening in tomato. Plant Physiol. 2016, 170, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Lund, S.T.; Stall, R.E.; Klee, H.J. Ethylene regulates the susceptible response to pathogen infection in tomato. Plant Cell 1998, 10, 371–382. [Google Scholar] [CrossRef]
- Wilkinson, J.Q.; Lanahan, M.B.; Yen, H.-C.; Giovannoni, J.J.; Klee, H.J. An ethylene-inducible component of signal transduction encoded by never-ripe. Science 1995, 270, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pirrello, J.; Chervin, C.; Roustan, J.P.; Bouzayen, M. Ethylene control of fruit ripening: Revisiting the complex network of transcriptional regulation. Plant Physiol. 2015, 169, 2380–2390. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Gu, Y.; Friman, V.P.; Kowalchuk, G.A.; Jousset, A. Initial soil microbiome composition and functioning predetermine future plant health. Sci. Adv. 2019, 5, eaaw0759. [Google Scholar] [CrossRef] [Green Version]
- Kuijken, R.C.P.; Snel, J.F.H.; Heddes, M.M.; Bouwmeester, H.J.; Marcelis, L.F.M. The importance of a sterile rhizosphere when phenotyping for root exudation. Plant Soil 2015, 387, 131–142. [Google Scholar] [CrossRef]
- Vranova, V.; Rejsek, K.; Skene, K.R.; Janous, D.; Formanek, P. Methods of collection of plant root exudates in relation to plant metabolism and purpose: A review. J. Plant Nutr. Soil Sci. 2013, 176, 175–199. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G.; Smith, C.A. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Poinar, H.N.; Schwarz, C.; Ji, Q.; Shapiro, B.; Macphee, R.; Buigues, B.; Tikhonov, A.; Huson, D.H.; Tomsho, L.P.; Auch, A. Metagenomics to Paleogenomics: Large-Scale Sequencing of Mammoth DNA. Science 2006, 311, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Warton, D.I.; Wright, S.T.; Wang, Y. Distance-based multivariate analyses confound location and dispersion effects. Methods Ecol. Evol. 2012, 3, 89–101. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: cran.r-project.org/package=vegan (accessed on 24 November 2021).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A language and environment for statistical computing. Computing 2014, 1, 12–21. [Google Scholar]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; Glavina del Rio, T.; Jones, C.D.; Tringe, S.G.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.C.; Jiang, T.; Liu, Y.-X.; Bai, Y.-C.; Reed, J.; Qu, B.; Goossens, A.; Nützmann, H.-W.; Bai, Y.; Osbourn, A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science 2019, 364, eaau6389. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Cha, J.-Y.; Han, S.; Hong, H.-J.; Cho, H.; Kim, D.; Kwon, Y.; Kwon, S.-K.; Crüsemann, M.; Bok Lee, Y.; Kim, J.F.; et al. Microbial and biochemical basis of a Fusarium wilt-suppressive soil. ISME J. 2016, 10, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichmann, R.; Richards, L.; Schäfer, P. Hormones as go-betweens in plant microbiome assembly. Plant J. 2021, 105, 518–541. [Google Scholar] [CrossRef]
- Bari, R.; Jones, J.D.G. Role of plant hormones in plant defence responses. Plant Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Badri, D.V.; Vivanco, J.M. Regulation and function of root exudates. Plant Cell Environ. 2009, 32, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; George, T.S.; Plassard, C. Strategies and methods for studying the rhizosphere-the plant science toolbox. Plant Soil 2009, 321, 431–456. [Google Scholar] [CrossRef]
- Williams, A.; Langridge, H.; Straathof, A.L.; Fox, G.; Muhammadali, H.; Hollywood, K.A.; Xu, Y.; Goodacre, R.; de Vries, F.T. Comparing root exudate collection techniques: An improved hybrid method. Soil Biol. Biochem. 2021, 161, 108391. [Google Scholar] [CrossRef]
- Oburger, E.; Dell‘mour, M.; Hann, S.; Wieshammer, G.; Puschenreiter, M.; Wenzel, W.W. Evaluation of a novel tool for sampling root exudates from soil-grown plants compared to conventional techniques. Environ. Exp. Bot. 2013, 87, 235–247. [Google Scholar] [CrossRef]
- Canarini, A.; Kaiser, C.; Merchant, A.; Richter, A.; Wanek, W. Corrigendum: Root exudation of primary metabolites: Mechanisms and their roles in plant responses to environmental stimuli. Front. Plant Sci. 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantigoso, H.A.; He, Y.; DiLegge, M.J.; Vivanco, J.M. Methods for Root Exudate Collection and Analysis. In The Plant Microbiome; Springer Nature: Berlin/Heidelberg, Germany, 2021; Volume 69, pp. 291–303. ISBN 9781071610398. [Google Scholar]
- Dombrowski, N.; Schlaeppi, K.; Agler, M.T.; Hacquard, S.; Schulze-Lefert, P. Root microbiota dynamics of perennial Arabis alpina are dependent on soil residence time but independent of flowering time. ISME J. 2016, 11, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, N.; Liu, Y.-X.; Zhang, X.; Hu, B.; Qin, Y.; Xu, H.; Wang, H.; Guo, X.; Qian, J.; et al. Root microbiota shift in rice correlates with resident time in the field and developmental stage. Sci. China Life Sci. 2018, 61, 613–621. [Google Scholar] [CrossRef]
- Stringlis, I.A.; Yu, K.; Feussner, K.; De Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.L.; Bakker, P.A.H.M.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voges, M.J.; Bai, Y.; Schulze-Lefert, P.; Sattely, E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 12558–12565. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Zhang, N.; Du, W.; Zhang, H.; Liu, Y.; Fu, R.; Shao, J.; Zhang, G.; Shen, Q.; Zhang, R. Identification of chemotaxis compounds in root exudates and their sensing chemoreceptors in plant-growth-promoting rhizobacteria Bacillus amyloliquefaciens SQR9. Mol. Plant-Microbe Interact. 2018, 31, 995–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Zhang, N.; Fu, R.; Liu, Y.; Krell, T.; Du, W.; Shao, J.; Shen, Q.; Zhang, R. Recognition of dominant attractants by key chemoreceptors mediates recruitment of plant growth-promoting rhizobacteria. Environ. Microbiol. 2019, 21, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; Friis, L.M.; Nothaft, H.; Liu, X.; Li, J.; Szymanski, C.M.; Stintzi, A. L-Fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc. Natl. Acad. Sci. USA 2011, 108, 7194–7199. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Ding, W.; Xu, Y.; Wu, D.; Li, S.; Chen, J.; Guo, B. New Insights into the Antibacterial Activity of Hydroxycoumarins against Ralstonia solanacearum. Molecules 2016, 21, 468. [Google Scholar] [CrossRef]
- Yuan, J.; Zhao, J.; Wen, T.; Zhao, M.; Li, R.; Goossens, P.; Huang, Q.; Bai, Y.; Vivanco, J.M.; Kowalchuk, G.A.; et al. Root exudates drive the soil-borne legacy of aboveground pathogen infection. Microbiome 2018, 6, 156. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, R.; Feng, H.; Dini-Andreote, F.; Wang, Z.; Bo, C.; Cao, W.; Yang, K.; Liu, M.; Yang, T.; Shen, Q.; et al. Modulation of the Tomato Rhizosphere Microbiome via Changes in Root Exudation Mediated by the Ethylene Receptor NR. Microorganisms 2021, 9, 2456. https://doi.org/10.3390/microorganisms9122456
Fu R, Feng H, Dini-Andreote F, Wang Z, Bo C, Cao W, Yang K, Liu M, Yang T, Shen Q, et al. Modulation of the Tomato Rhizosphere Microbiome via Changes in Root Exudation Mediated by the Ethylene Receptor NR. Microorganisms. 2021; 9(12):2456. https://doi.org/10.3390/microorganisms9122456
Chicago/Turabian StyleFu, Ruixin, Haichao Feng, Francisco Dini-Andreote, Zhen Wang, Chunbin Bo, Wenhui Cao, Keming Yang, Mingchun Liu, Tianjie Yang, Qirong Shen, and et al. 2021. "Modulation of the Tomato Rhizosphere Microbiome via Changes in Root Exudation Mediated by the Ethylene Receptor NR" Microorganisms 9, no. 12: 2456. https://doi.org/10.3390/microorganisms9122456
APA StyleFu, R., Feng, H., Dini-Andreote, F., Wang, Z., Bo, C., Cao, W., Yang, K., Liu, M., Yang, T., Shen, Q., Xu, Y., & Wei, Z. (2021). Modulation of the Tomato Rhizosphere Microbiome via Changes in Root Exudation Mediated by the Ethylene Receptor NR. Microorganisms, 9(12), 2456. https://doi.org/10.3390/microorganisms9122456