
Diversity Distribution, Driving Factors and Assembly Mechanisms of Free-Living and Particle-Associated Bacterial Communities at a Subtropical Marginal Sea


 , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
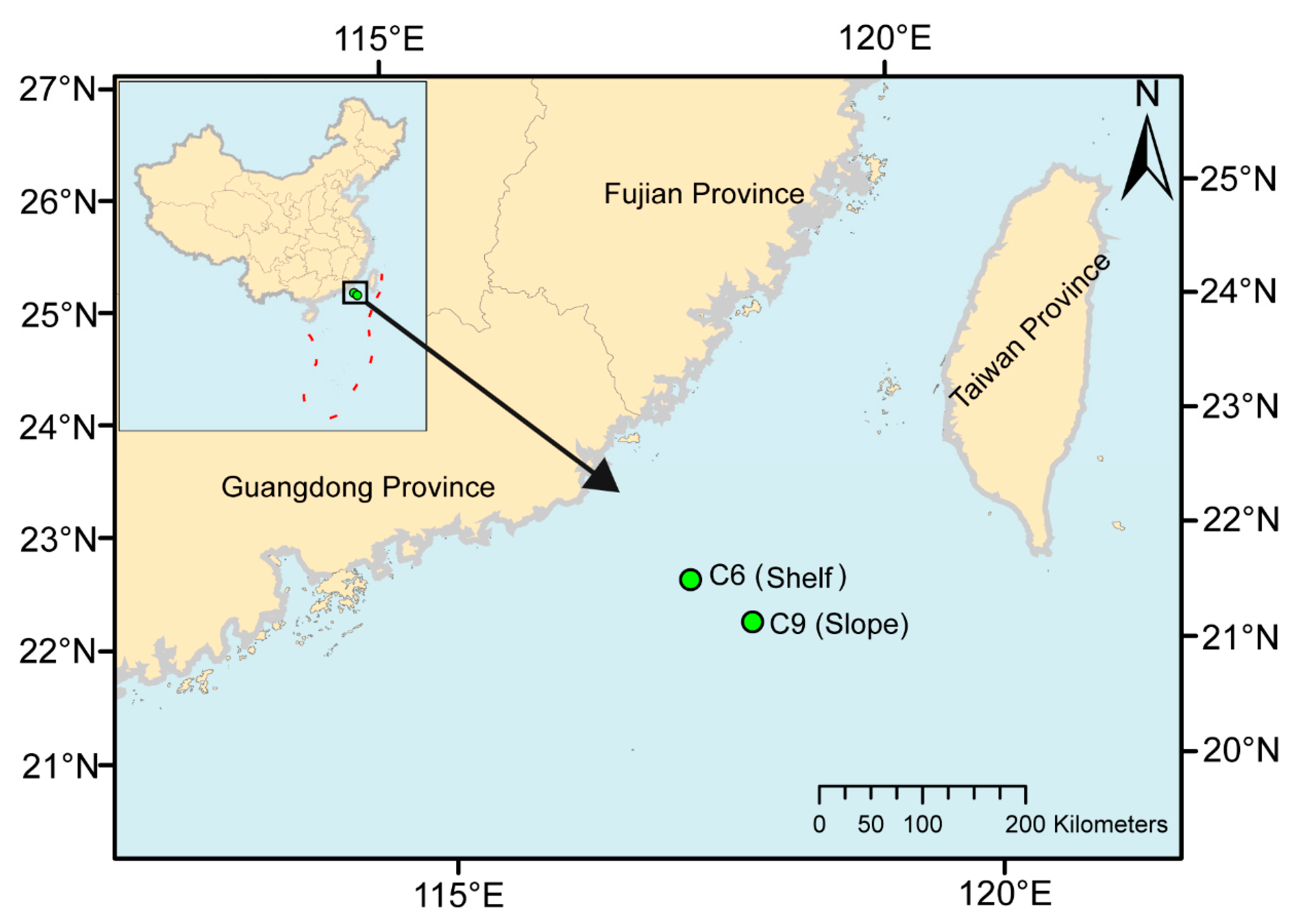
2.1. Sample Collection and Environmental Variables
2.2. DNA Extraction and Sequencing
2.3. DNA Sequence Analysis and Function Inference
2.4. Quantification of Community Assembly Processes
2.5. Statistical Analyses
3. Results
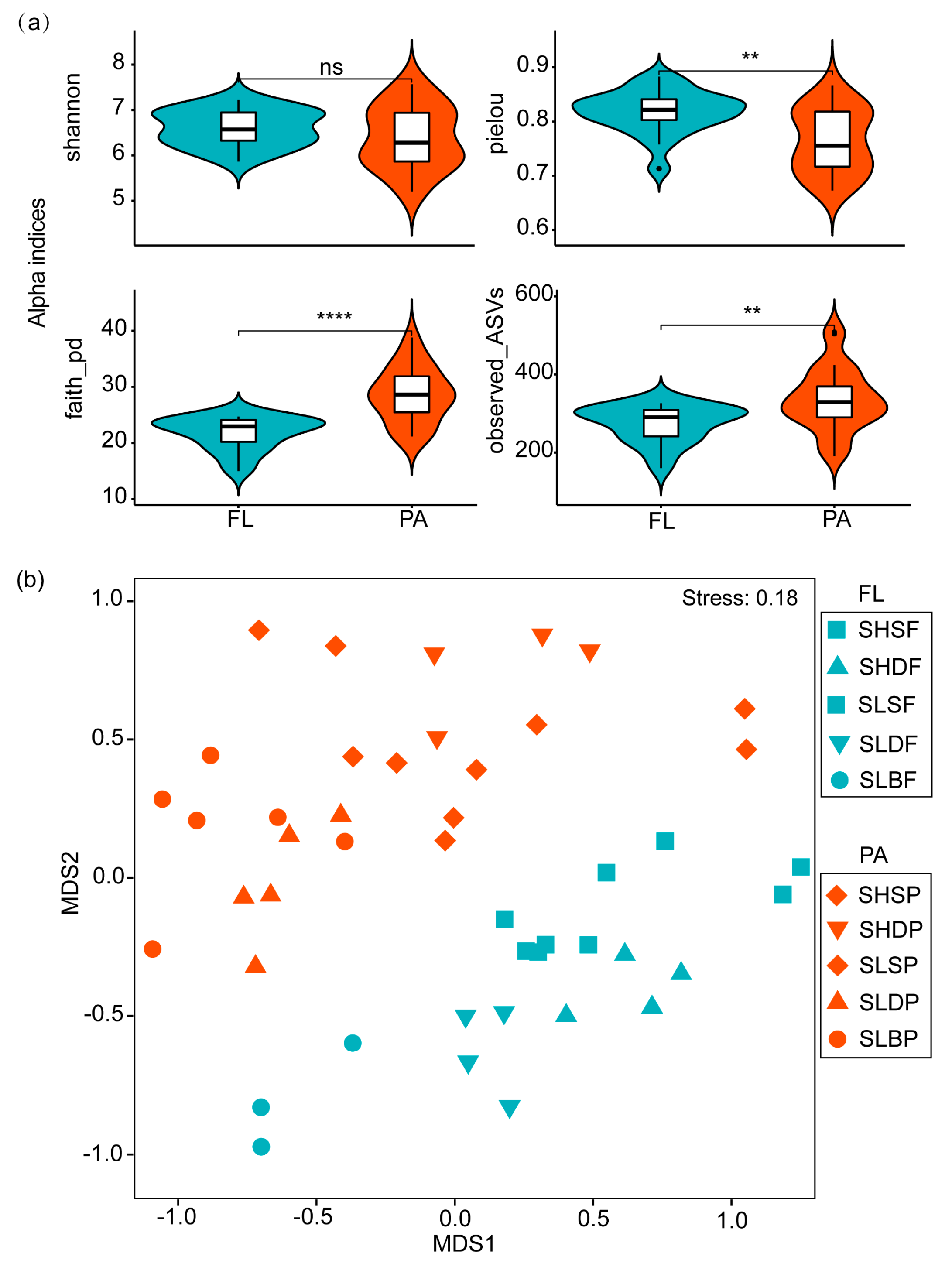
3.1. Spatial Variations in Alpha and Beta Diversity
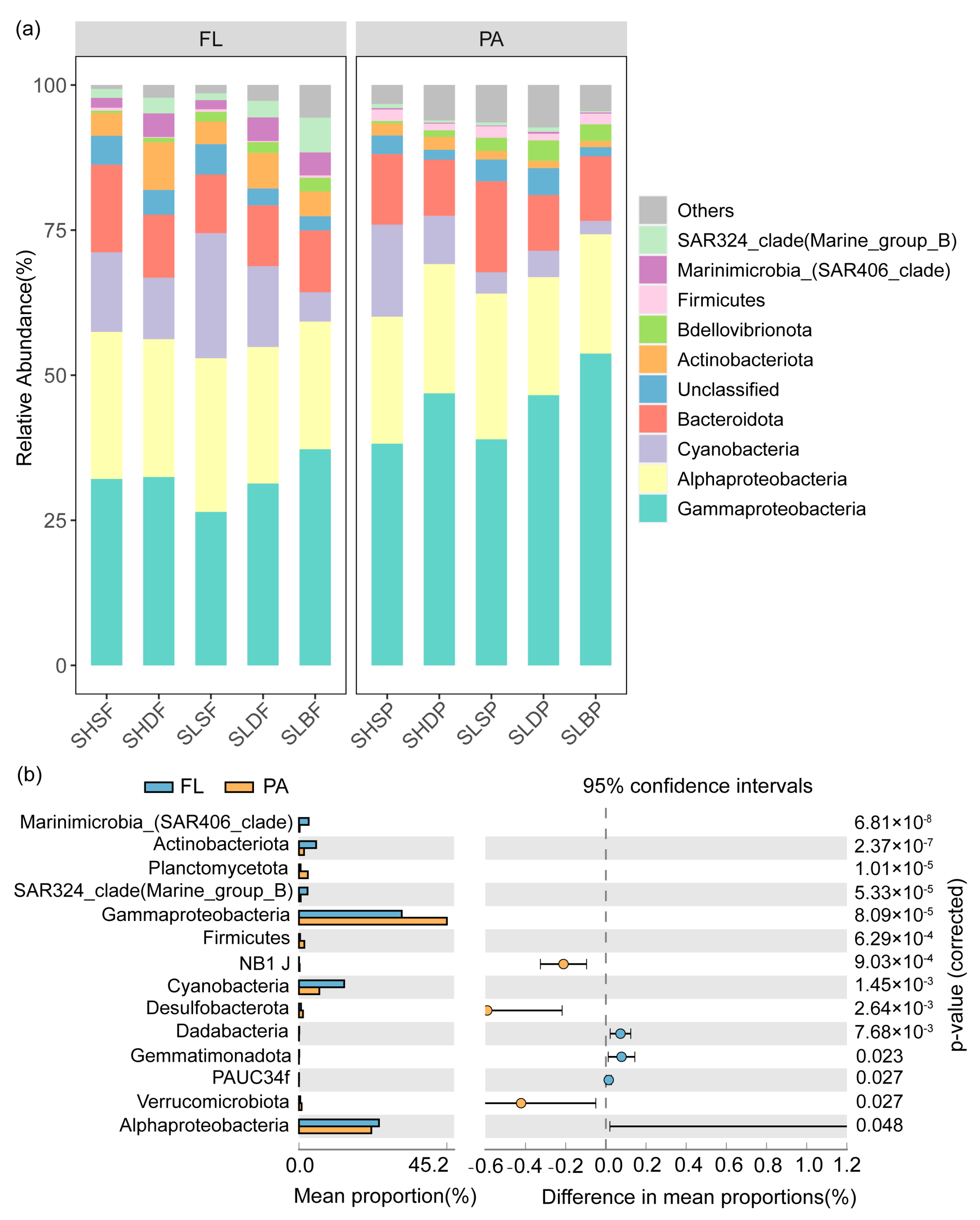
3.2. Spatial Variation of Bacterioplankton Community
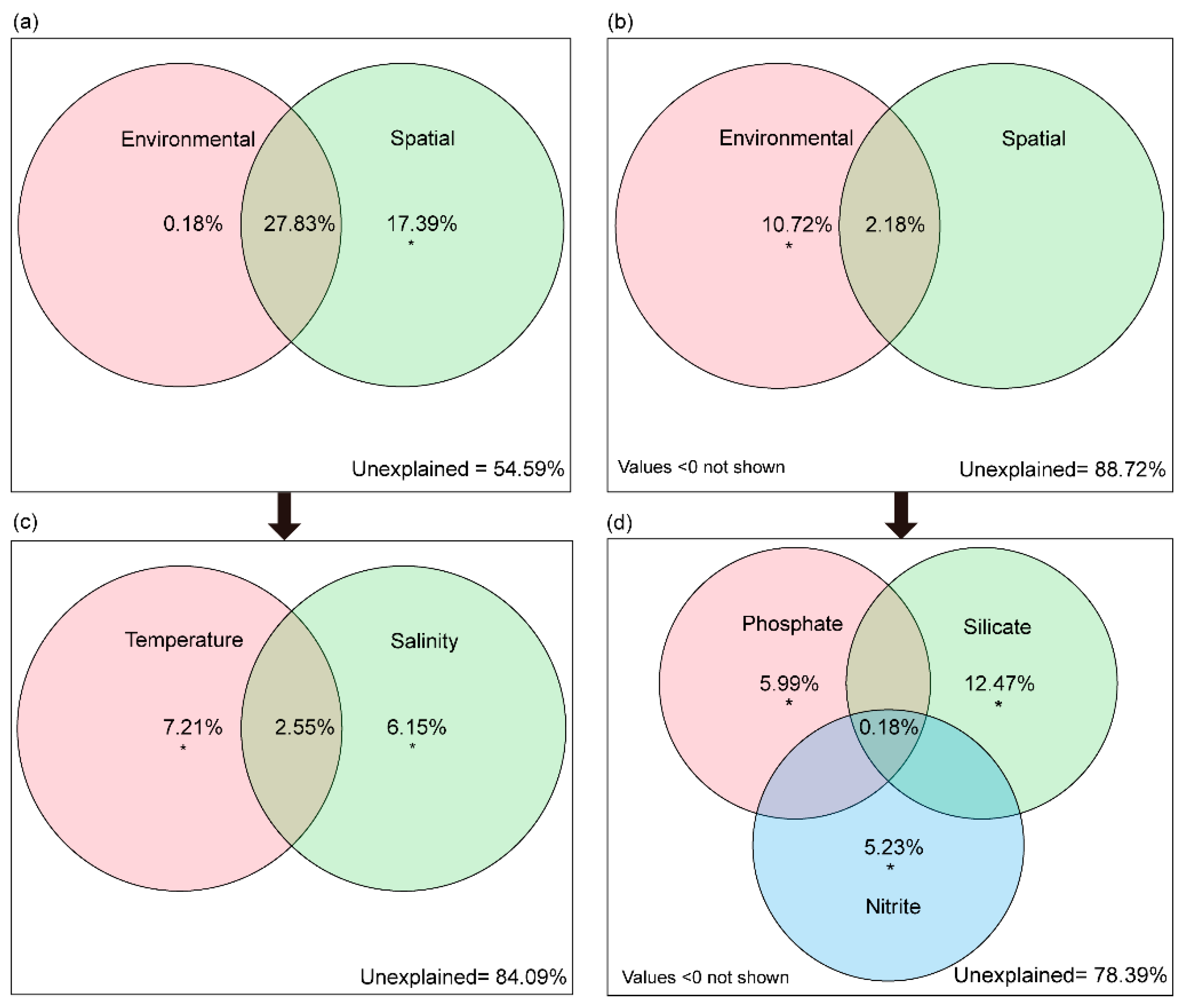
3.3. Environmental and Spatial Factors Influencing Bacterioplankton Community
3.4. Community Assembly Mechanisms of Bacterioplankton
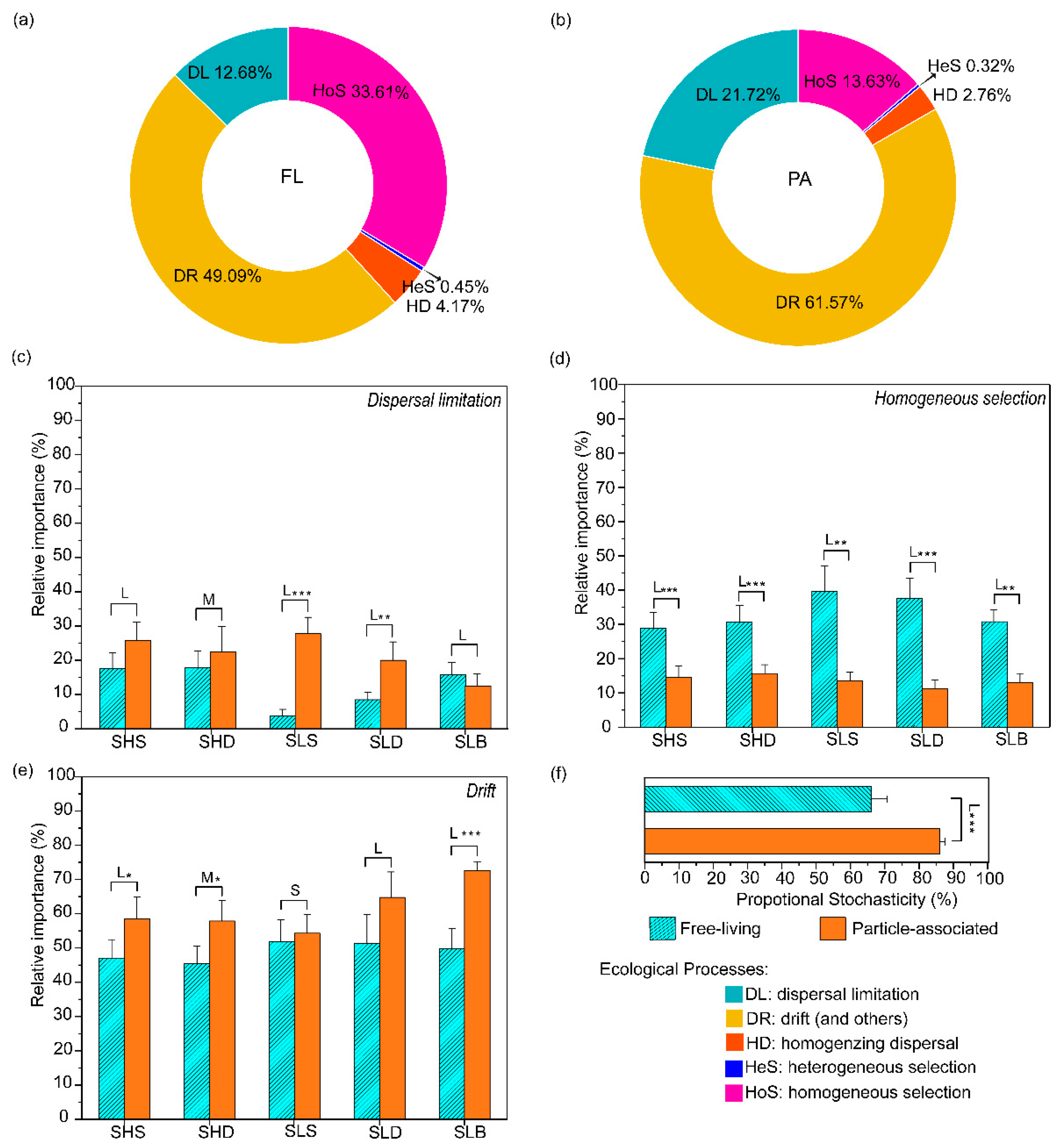
3.4.1. Effects of Lifestyle on Bacterioplankton Community Assembly
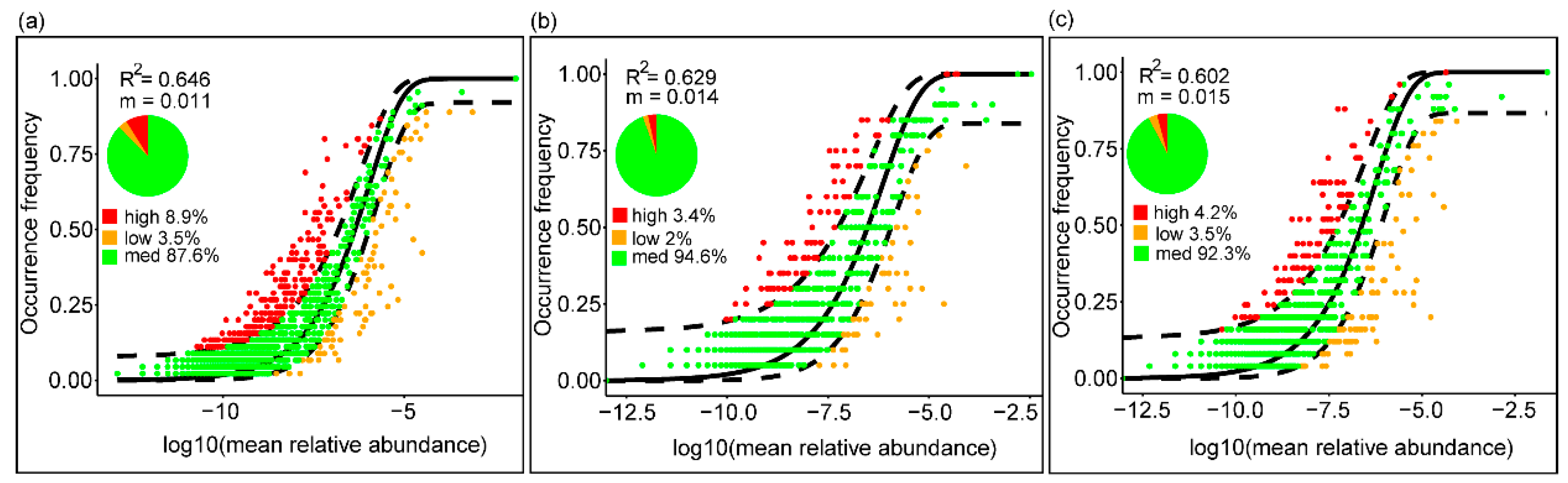
3.4.2. Stochastic vs. Deterministic Bacterial Assembly
3.4.3. Assembly Mechanisms across Different Phylogenetic Groups
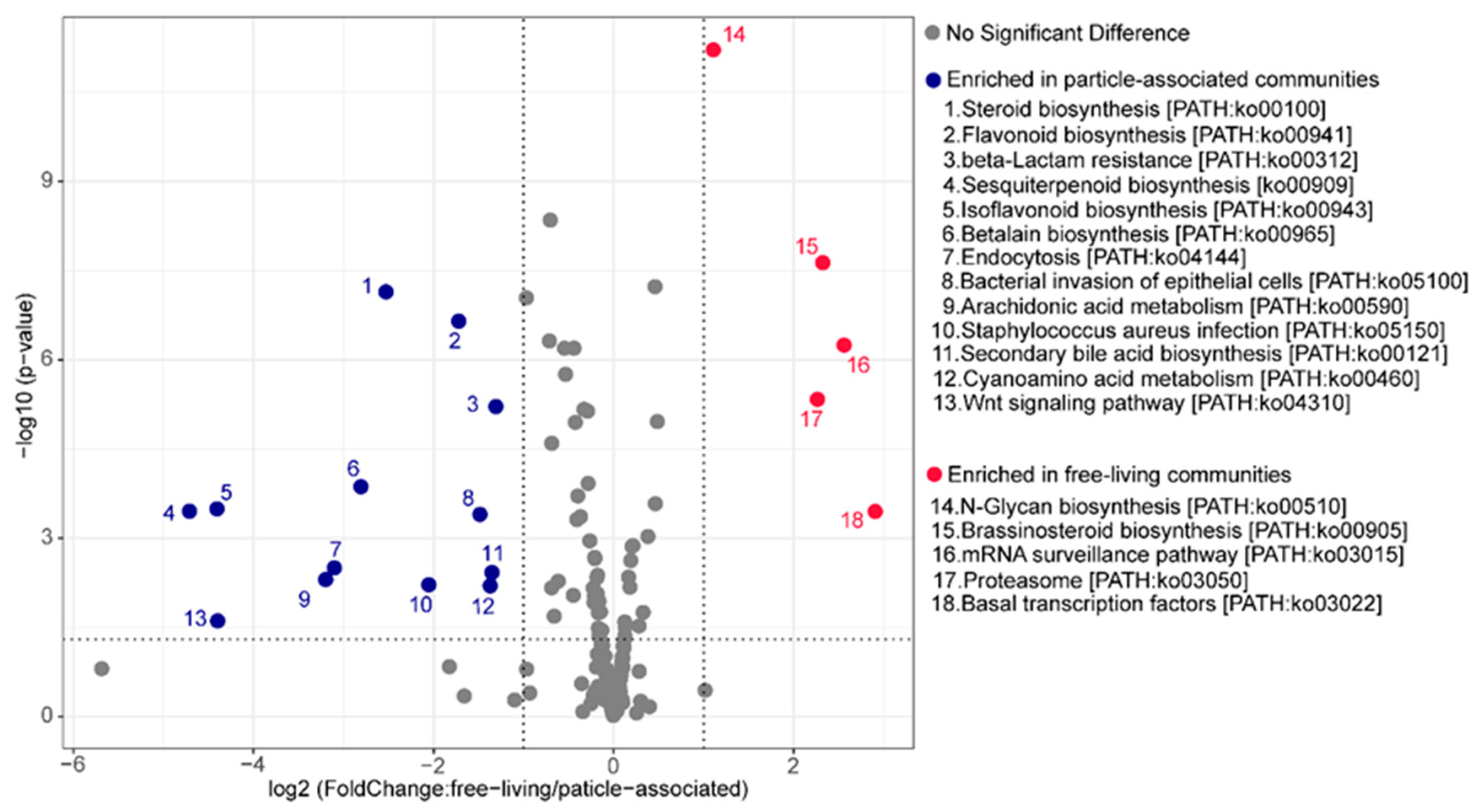
3.5. Predicting Metabolic Functions That Might Underlie Changes between the FL and PA Community Structure
4. Discussion
4.1. General Structure of Bacterioplankton Community Structure across Size Fraction, Water Depth and Station
4.2. Different Environmental Factors Affecting the FL and PA Bacterioplankton Communities
4.3. Stochastic Processes Dominated Bacterioplankton Community Assembly
4.4. Predicted Metabolic Potential in the FL and PA Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freimann, R.; Bürgmann, H.; Findlay, S.E.; Robinson, C.T. Bacterial structures and ecosystem functions in glaciated floodplains: Contemporary states and potential future shifts. ISME J. 2013, 7, 2361–2373. [Google Scholar] [CrossRef] [Green Version]
- Lopes Dos Santos, A.; Gourvil, P.; Tragin, M.; Noel, M.H.; Decelle, J.; Romac, S.; Vaulot, D. Diversity and oceanic distribution of prasinophytes clade VII, the dominant group of green algae in oceanic waters. ISME J. 2017, 11, 512–528. [Google Scholar] [CrossRef] [Green Version]
- Gutknecht, J.L.M.; Field, C.B.; Balser, T.C. Microbial communities and their responses to simulated global change fluctuate greatly over multiple years. Glob. Chang. Biol. 2012, 18, 2256–2269. [Google Scholar] [CrossRef]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Genet. 2007, 5, 782–791. [Google Scholar] [CrossRef]
- Guidi, L.; Chaffron, S.; Bittner, L.; Eveillard, D.; Larhlimi, A.; Roux, S.; Darzi, Y.; Audic, S.; Berline, L.; Brum, J.R.; et al. Plankton networks driving carbon export in the oligotrophic ocean. Nature 2016, 532, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Letscher, R.T.; Knapp, A.N.; James, A.K.; Carlson, C.A.; Santoro, A.; Hansell, D. Microbial community composition and nitrogen availability influence DOC remineralization in the South Pacific Gyre. Mar. Chem. 2015, 177, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [Green Version]
- Azam, F.; Long, R.A. Sea snow microcosms. Nature 2001, 414, 497–498. [Google Scholar] [CrossRef] [PubMed]
- Aristegui, J.; Gasol, J.M.; Duarte, C.M.; Herndl, G.J. Microbial oceanography of the dark ocean’s pelagic realm. Limnol. Oceanogr. 2009, 54, 1501–1529. [Google Scholar] [CrossRef] [Green Version]
- Karl, D.M. Microbial oceanography: Paradigms, processes and promise. Nat. Rev. Genet. 2007, 5, 759–769. [Google Scholar] [CrossRef]
- Mestre, M.; Borrull, E.; Sala, M.M.; Gasol, J.M. Patterns of bacterial diversity in the marine planktonic particulate matter continuum. ISME J. 2017, 11, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Moran, M.A. How do divergent ecological strategies emerge among marine bacterioplankton lineages? Trends Microbiol. 2015, 23, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Bižić-Ionescu, M.; Amann, R.; Grossart, H.-P. Massive Regime Shifts and High Activity of Heterotrophic Bacteria in an Ice-Covered Lake. PLoS ONE 2014, 9, e113611. [Google Scholar] [CrossRef]
- Tang, X.; Chao, J.; Gong, Y.; Wang, Y.; Wilhelm, S.; Gao, G. Spatiotemporal dynamics of bacterial community composition in large shallow eutrophic Lake Taihu: High overlap between free-living and particle-attached assemblages. Limnol. Oceanogr. 2017, 62, 1366–1382. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, D.; Huang, R.; Cao, X.; Zeng, J.; Yu, Z.; Hooker, K.V.; Hambright, K.D.; Wu, Q.L. Contrasting Network Features between Free-Living and Particle-Attached Bacterial Communities in Taihu Lake. Microb. Ecol. 2018, 76, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Savio, D.; Sinclair, L.; Ijaz, U.Z.; Parajka, J.; Reischer, G.; Stadler, P.; Blaschke, A.P.; Blöschl, G.; Mach, R.; Kirschner, A.; et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 2015, 17, 4994–5007. [Google Scholar] [CrossRef]
- Henson, M.W.; Hanssen, J.; Spooner, G.; Fleming, P.; Pukonen, M.; Stahr, F.; Thrash, J.C. Nutrient dynamics and stream order influence microbial community patterns along a 2914 kilometer transect of the Mississippi River. Limnol. Oceanogr. 2018, 63, 1837–1855. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, K.; York, J.; Biddle, J.F. Impacts of Salinity and Oxygen on Particle-Associated Microbial Communities in the Broadkill River, Lewes DE. Front. Mar. Sci. 2018, 5, 100. [Google Scholar] [CrossRef]
- Smith, M.W.; Allen, L.Z.; Allen, A.E.; Herfort, L.; Simon, H.M. Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem. Front. Microbiol. 2013, 4, 120. [Google Scholar] [CrossRef] [Green Version]
- Bižić-Ionescu, M.; Zeder, M.; Ionescu, D.; Orlic, S.; Fuchs, B.; Grossart, H.-P.; Amann, R. Comparison of bacterial communities on limnic versus coastal marine particles reveals profound differences in colonization. Environ. Microbiol. 2014, 17, 3500–3514. [Google Scholar] [CrossRef] [PubMed]
- Rieck, A.; Herlemann, D.; Jürgens, K.; Grossart, H.-P. Particle-Associated Differ from Free-Living Bacteria in Surface Waters of the Baltic Sea. Front. Microbiol. 2015, 6, 1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, M.; Yager, P.; Moran, M.A.; Coles, V.J.; Fortunato, C.S.; Krusche, A.; Medeiros, P.M.; Payet, J.P.; Richey, J.E.; Satinsky, B.; et al. Bacterial Biogeography across the Amazon River-Ocean Continuum. Front. Microbiol. 2017, 8, 882. [Google Scholar] [CrossRef]
- Kieft, B.; Li, Z.; Bryson, S.; Crump, B.C.; Hettich, R.; Pan, C.; Mayali, X.; Mueller, R.S. Microbial Community Structure–Function Relationships in Yaquina Bay Estuary Reveal Spatially Distinct Carbon and Nitrogen Cycling Capacities. Front. Microbiol. 2018, 9, 1282. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Luo, H.; Murugapiran, S.K.; Dodsworth, J.A.; Chen, S.; Sun, Y.; Hedlund, B.P.; Wang, P.; Fang, H.; Deng, M.; et al. Localized high abundance of Marine Group II archaea in the subtropical Pearl River Estuary: Implications for their niche adaptation. Environ. Microbiol. 2018, 20, 734–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, J.; Yang, J.; Zhou, Z.; Pan, Y.; Li, M. Patterns and processes of free-living and particle-associated bacterioplankton and archaeaplankton communities in a subtropical river-bay system in South China. Limnol. Oceanogr. 2020, 65. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Cornejo-Castillo, F.M.; Benítez-Barrios, V.; Fraile-Nuez, E.; Alvarez-Salgado, X.A.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 2016, 10, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Milici, M.; Deng, Z.L.; Tomasch, J.; Decelle, J.; Wos-Oxley, M.L.; Wang, H.; Jauregui, R.; Plumeier, I.; Giebel, H.A.; Badewien, T.H.; et al. Co-occurrence Analysis of Microbial Taxa in the Atlantic Ocean Reveals High Connectivity in the Free-Living Bacterioplankton. Front. Microbiol. 2016, 7, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, E.A.; Pachiadaki, M.; Taylor, G.T.; Astor, Y.; Edgcomb, V.P. Free-living chemoautotrophic and particle-attached heterotrophic prokaryotes dominate microbial assemblages along a pelagic redox gradient. Environ. Microbiol. 2018, 20, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.; Hoffmann, L.J.; Larcombe, M.J.; Louisson, Z.; Summerfield, T.C. Homogeneous environmental selection dominates microbial community assembly in the oligotrophic South Pacific Gyre. Mol. Ecol. 2020, 29, 4680–4691. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Meng, Z.; Liu, X.; Zhang, X.-H. Microbial assembly, interaction, functioning, activity and diversification: A review derived from community compositional data. Mar. Life Sci. Technol. 2019, 1, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [Green Version]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and Processes of Microbial Community Assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, J.M.; Leibold, M.A. Ecological Niches: Linking Classical and Contemporary Approaches; University of Chicago Press: Chicago, IL, USA, 2004; Volume 13, pp. 1791–1793. [Google Scholar]
- Jetschke, G. The Unified Neutral Theory of Biodiversity and Biogeography. Ecology 2002, 83, 1771–1772. [Google Scholar] [CrossRef]
- Vellend, M. Conceptual Synthesis in Community Ecology. Q. Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef] [Green Version]
- Vellend, M.; Srivastava, D.S.; Anderson, K.M.; Brown, C.D.; Jankowski, J.E.; Kleynhans, E.J.; Kraft, N.; Letaw, A.D.; Macdonald, A.A.M.; Maclean, J.E.; et al. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 2014, 123, 1420–1430. [Google Scholar] [CrossRef]
- Vellend, M. The Theory of Ecological Communities; Princeton University Press: Princeton, NJ, USA, 2016. [Google Scholar]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 2020, 11, 4717. [Google Scholar] [CrossRef]
- Stegen, J.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Stegen, J.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Lu, H.-P.; Sastri, A.; Yeh, Y.-C.; Gong, G.-C.; Chou, W.-C.; Hsieh, C.-H. Contrasting the relative importance of species sorting and dispersal limitation in shaping marine bacterial versus protist communities. ISME J. 2017, 12, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Mo, Y.; Zhang, W.; Yang, J.; Lin, Y.; Yu, Z.; Lin, S. Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J. 2018, 12, 2198–2210. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Yan, H.; Peng, X.; Hu, H.; Zhang, H.; Hou, D.; Chen, W.; Qian, P.; Liu, J.; Cai, J.; et al. Community assembly of bacteria and archaea in coastal waters governed by contrasting mechanisms: A seasonal perspective. Mol. Ecol. 2020, 29, 3762–3776. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Song, X.; He, D.; Wang, J.; Tan, M.; Xia, X.; Li, G.; Tan, Y.; Wu, Q.L. Bacterioplankton Metacommunity Processes across Thermal Gradients: Weaker Species Sorting but Stronger Niche Segregation in Summer than in Winter in a Subtropical Bay. Appl. Environ. Microbiol. 2019, 85, e02088-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollenhauer, G.; Schneider, R.; Müller, P.J.; Spiess, V.; Wefer, G. Glacial/interglacial variablity in the Benguela upwelling system: Spatial distribution and budgets of organic carbon accumulation. Glob. Biogeochem. Cycles 2002, 16, 81-1–81-15. [Google Scholar] [CrossRef]
- Satinsky, B.; Crump, B.; Smith, C.B.; Sharma, S.; Zielinski, B.L.; Doherty, M.; Meng, J.; Sun, S.; Medeiros, P.M.; Paul, J.H.; et al. Microspatial gene expression patterns in the Amazon River Plume. Proc. Natl. Acad. Sci. USA 2014, 111, 11085–11090. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.M.; Smith, M.W.; Herfort, L. Metagenomic insights into particles and their associated microbiota in a coastal margin ecosystem. Front. Microbiol. 2014, 5, 466. [Google Scholar] [CrossRef]
- Yuan, J.; Li, M.; Lin, S. An Improved DNA Extraction Method for Efficient and Quantitative Recovery of Phytoplankton Diversity in Natural Assemblages. PLoS ONE 2015, 10, e0133060. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Caicedo, H.H.; Hashimoto, D.A.; Caicedo, J.C.; Pentland, A.; Pisano, G.P. Overcoming barriers to early disease intervention. Nat. Biotechnol. 2020, 38, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Logares, R.; Lindström, E.; Langenheder, S.; Logue, J.B.; Paterson, H.; Laybourn-Parry, J.; Rengefors, K.; Tranvik, L.J.; Bertilsson, S. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J. 2012, 7, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Cao, X.; Wang, J.; Zhao, L.; Sun, J.; Jiang, D.; Huang, Y. Similar community assembly mechanisms underlie similar biogeography of rare and abundant bacteria in lakes on Yungui Plateau, China. Limnol. Oceanogr. 2017, 62, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Chen, J.; van Oosterhout, C.; Zhang, H.; Liu, G.; Zhuang, Y.; Mock, T. Diversity, prevalence, and expression of cyanase genes (cynS) in planktonic marine microorganisms. ISME J. 2021, 1–4. [Google Scholar] [CrossRef]
- Ghiglione, J.F.; Mevel, G.; Pujo-Pay, M.; Mousseau, L.; Lebaron, P.; Goutx, M. Diel and Seasonal Variations in Abundance, Activity, and Community Structure of Particle-Attached and Free-Living Bacteria in NW Mediterranean Sea. Microb. Ecol. 2007, 54, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Mestre, M.; González, C.R.; Logares, R.; Duarte, C.M.; Gasol, J.M.; Sala, M.M. Sinking particles promote vertical connectivity in the ocean microbiome. Proc. Natl. Acad. Sci. USA 2018, 115, E6799–E6807. [Google Scholar] [CrossRef] [Green Version]
- Fontanez, K.M.; Eppley, J.M.; Samo, T.; Karl, D.; Delong, E.F. Microbial community structure and function on sinking particles in the North Pacific Subtropical Gyre. Front. Microbiol. 2015, 6, 469. [Google Scholar] [CrossRef] [Green Version]
- Haro-Moreno, J.M.; Lopez-Perez, M.; de la Torre, J.R.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Fine metagenomic profile of the Mediterranean stratified and mixed water columns revealed by assembly and recruitment. Microbiome 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, C.-M.; Ward, C.S.; Davis, K.M.; Johnson, Z.I.; Hunt, D.E. Insensitivity of Diverse and Temporally Variable Particle-Associated Microbial Communities to Bulk Seawater Environmental Parameters. Appl. Environ. Microbiol. 2016, 82, 3431–3437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, J.; Heimbach, T.; Hassenrück, C.; Kopprio, G.; Iversen, M.H.; Grossart, H.P.; Gärdes, A. Environmental Drivers of Free-Living vs. Particle-Attached Bacterial Community Composition in the Mauritania Upwelling System. Front. Microbiol. 2018, 9, 2836. [Google Scholar] [CrossRef]
- Roth Rosenberg, D.; Haber, M.; Goldford, J.; Lalzar, M.; Aharonovich, D.; Al-Ashhab, A.; Lehahn, Y.; Segre, D.; Steindler, L.; Sher, D. Particle-associated and free-living bacterial communities in an oligotrophic sea are affected by different environmental factors. Environ. Microbiol. 2021, 23, 4295–4308. [Google Scholar] [CrossRef]
- Langenheder, S.; Lindström, E.S. Factors influencing aquatic and terrestrial bacterial community assembly. Environ. Microbiol. Rep. 2019, 11, 306–315. [Google Scholar] [CrossRef]
- Roguet, A.; Laigle, G.S.; Therial, C.; Bressy, A.; Soulignac, F.; Catherine, A.; Lacroix, G.; Jardillier, L.; Bonhomme, C.; Lerch, T.Z.; et al. Neutral community model explains the bacterial community assembly in freshwater lakes. FEMS Microbiol. Ecol. 2015, 91, fiv125. [Google Scholar] [CrossRef]
- Burns, A.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.; Guillemin, K.; Bohannan, B.J. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A.; Cram, J.; Needham, D.M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Genet. 2015, 13, 133–146. [Google Scholar] [CrossRef]
- Bižić-Ionescu, M.; Ionescu, D.; Grossart, H.-P. Organic Particles: Heterogeneous Hubs for Microbial Interactions in Aquatic Ecosystems. Front. Microbiol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef] [Green Version]
- Meyerhof, M.S.; Wilson, J.M.; Dawson, M.N.; Beman, J.M. Microbial community diversity, structure and assembly across oxygen gradients in meromictic marine lakes, Palau. Environ. Microbiol. 2016, 18, 4907–4919. [Google Scholar] [CrossRef]
- Bracken, L.; Wainwright, J.; Ali, G.; Tetzlaff, D.; Smith, M.; Reaney, S.M.; Roy, A. Concepts of hydrological connectivity: Research approaches, pathways and future agendas. Earth-Sci. Rev. 2013, 119, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Grossart, H.-P.; Tang, K.W. www.aquaticmicrobial.net. Commun. Integr. Biol. 2010, 3, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, L.; Chen, H.; Yu, Z.; Yang, J.R.; Xue, Y.; Huang, B.; Yang, J. Community dynamics of free-living and particle-attached bacteria following a reservoir Microcystis bloom. Sci. Total Environ. 2019, 660, 501–511. [Google Scholar] [CrossRef]
- Ganesh, S.; Parris, D.J.; Delong, E.F.; Stewart, F.J. Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone. ISME J. 2013, 8, 187–211. [Google Scholar] [CrossRef] [Green Version]
- Von Rosen, T.; Keller, L.M.L.; Weber-Ban, E. Survival in Hostile Conditions: Pupylation and the Proteasome in Actinobacterial Stress Response Pathways. Front. Mol. Biosci. 2021, 8, 685757. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Process. | Definition | βNRI | RC |
|---|---|---|---|
| Homogeneous selection | Consistent selective pressure resulting from consistent environmental conditions is the primary cause of compositional turnover between a pair of local communities | <−1.96 | |
| Heterogeneous selection | Divergent selective pressure resulting from divergent environmental conditions is the primary cause of compositional turnover between a pair of local communities | >+1.96 | |
| Homogenizing dispersal | High dispersal rates between a pair of local communities is the primary cause of compositional turnover between a pair of local communities | <|1.96| | <−0.95 |
| Dispersal limitation | Low dispersal rates between a pair of local communities, acting in concert with ecological drift, are the primary cause of compositional turnover between a pair of local communities | <|1.96| | >+0.95 |
| Drift (and others) | Neither selection nor dispersal is the dominant process driving compositional turnover between a pair of local communities | <|1.96| | <|0.95| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, H.; Li, T.; Li, H.; Wang, C.; Li, L.; Lin, X.; Lin, S. Diversity Distribution, Driving Factors and Assembly Mechanisms of Free-Living and Particle-Associated Bacterial Communities at a Subtropical Marginal Sea. Microorganisms 2021, 9, 2445. https://doi.org/10.3390/microorganisms9122445
Yuan H, Li T, Li H, Wang C, Li L, Lin X, Lin S. Diversity Distribution, Driving Factors and Assembly Mechanisms of Free-Living and Particle-Associated Bacterial Communities at a Subtropical Marginal Sea. Microorganisms. 2021; 9(12):2445. https://doi.org/10.3390/microorganisms9122445
Chicago/Turabian StyleYuan, Huatao, Tangcheng Li, Hongfei Li, Cong Wang, Ling Li, Xin Lin, and Senjie Lin. 2021. "Diversity Distribution, Driving Factors and Assembly Mechanisms of Free-Living and Particle-Associated Bacterial Communities at a Subtropical Marginal Sea" Microorganisms 9, no. 12: 2445. https://doi.org/10.3390/microorganisms9122445
APA StyleYuan, H., Li, T., Li, H., Wang, C., Li, L., Lin, X., & Lin, S. (2021). Diversity Distribution, Driving Factors and Assembly Mechanisms of Free-Living and Particle-Associated Bacterial Communities at a Subtropical Marginal Sea. Microorganisms, 9(12), 2445. https://doi.org/10.3390/microorganisms9122445