
Phylogenetic Analysis Reveals Source Attribution Patterns for Campylobacter spp. in Tennessee and Pennsylvania

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolates and Sequencing
2.2. Phylogenetic Analysis and Taxonomic Identification
2.3. Cluster Identification
3. Results
3.1. Most Isolates Were Identified as C. jejuni and C. coli
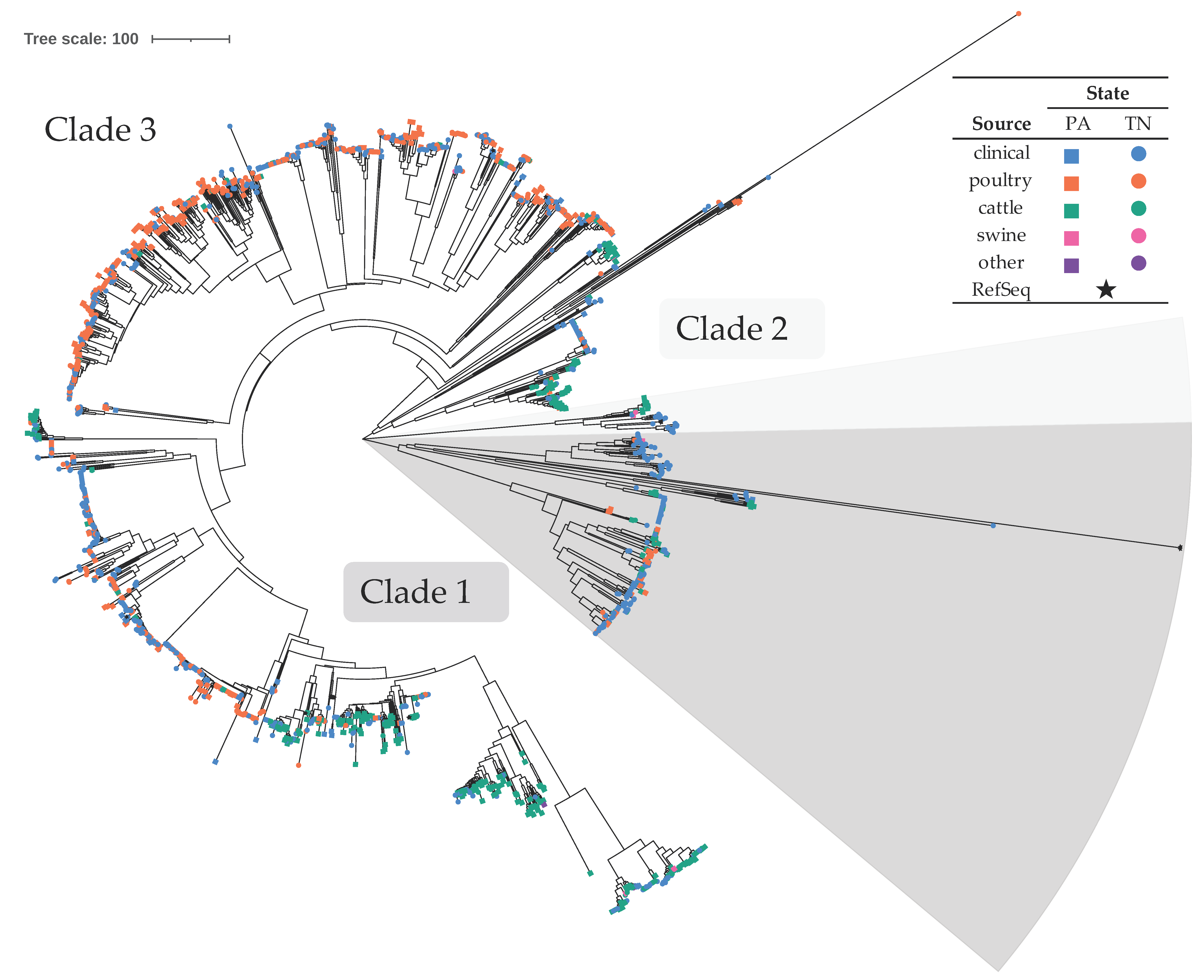
3.2. C. jejuni Formed Three Clades, Primarily Consisting of Poultry and Bovine Isolates
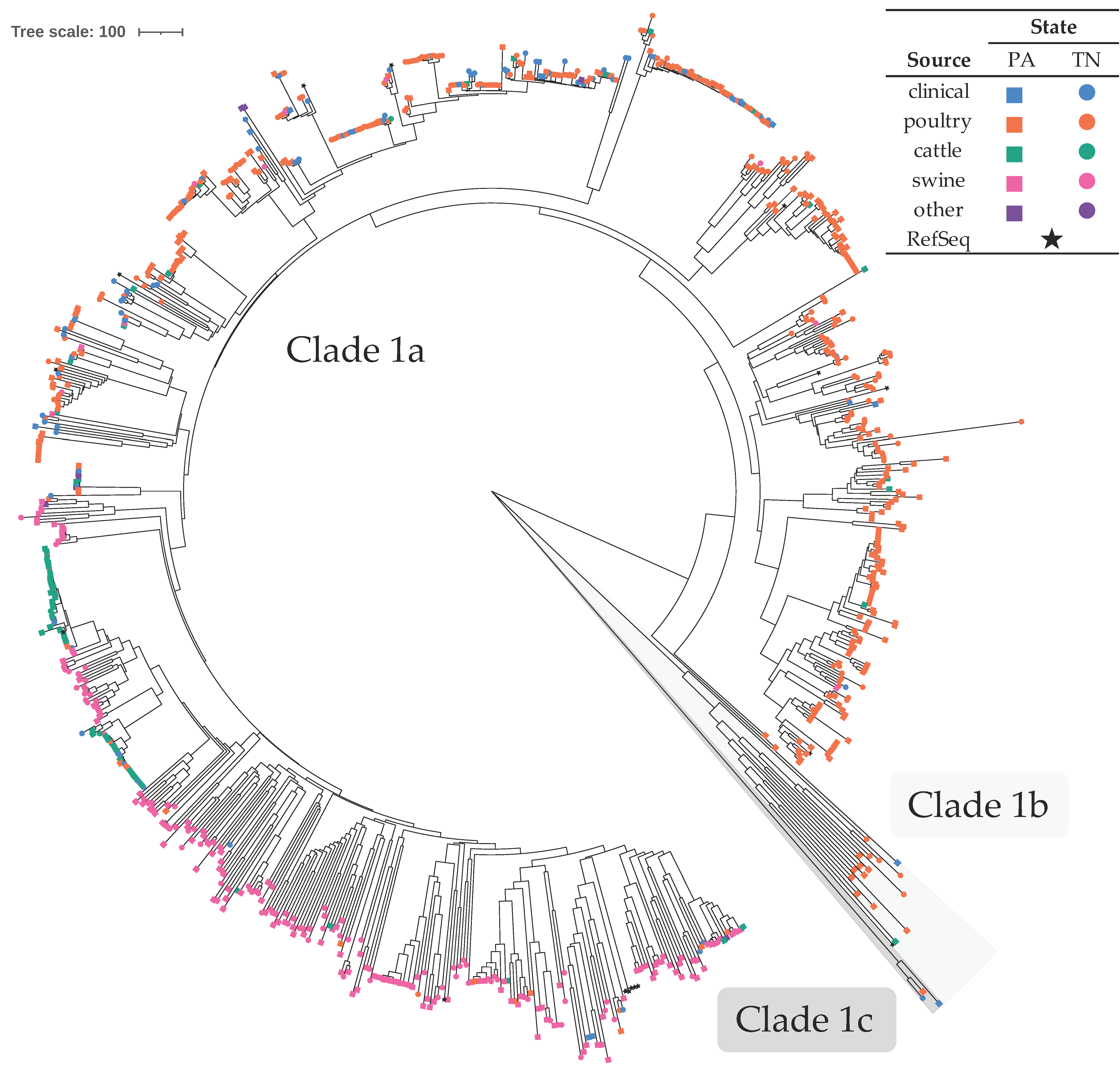
3.3. C. coli Formed Two Clades, Primarily Consisting of Poultry and Swine Isolates
3.4. Other Species
3.5. Potential Outbreak Clusters
4. Discussion
4.1. Source Attribution Trends
4.1.1. C. jejuni Associated with Poultry and Bovine Sources
4.1.2. C. coli Associated with Poultry, Swine, and Bovine Sources
4.1.3. Other Campylobacter spp.
4.2. Higher Proportion of Clinical Campylobacter Isolates May Be Outbreak-Associated
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tack, D.M.; Ray, L.; Griffin, P.M.; Cieslak, P.R.; Dunn, J.; Rissman, T.; Jervis, R.; Lathrop, S.; Muse, A.; Duwell, M. Preliminary incidence and trends of infections with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 US Sites, 2016–2019. Morb. Mortal. Wkly. Rep. 2020, 69, 509. [Google Scholar] [CrossRef]
- U.S. Department of Agriculture (USDA), E.R.S.E. Cost Estimates of Foodborne Illnesses. Available online: https://www.ers.usda.gov/data-products/cost-estimates-of-foodborne-illnesses/ (accessed on 16 August 2021).
- Centers for Disease Control and Prevention (CDC). Campylobacter (Campylobacteriosis): Questions and Answers. Available online: https://www.cdc.gov/campylobacter/faq.html (accessed on 28 January 2021).
- Keithlin, J.; Sargeant, J.; Thomas, M.K.; Fazil, A. Systematic review and meta-analysis of the proportion of Campylobacter cases that develop chronic sequelae. BMC Public Health 2014, 14, 1203. [Google Scholar] [CrossRef]
- Parte, A.C. LPSN—List of Prokaryotic names with Standing in Nomenclature (bacterio.net), 20 years on. Int. J. Syst. Evol. Microbiol. 2018, 68, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- LPSN. Genus Campylobacter. Available online: https://lpsn.dsmz.de/genus/campylobacter (accessed on 13 April 2021).
- Centers for Disease Control and Prevention (CDC). Foodborne Diseases Active Surveillance Network (FoodNet): FoodNet 2015 Surveillance Report (Final Data). Available online: https://www.cdc.gov/foodnet/pdfs/FoodNet-Annual-Report-2015-508c.pdf (accessed on 28 January 2021).
- Igwaran, A.; Okoh, A.I. Human campylobacteriosis: A public health concern of global importance. Heliyon 2019, 5, e02814. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ma, R.N.; Wang, Y.M.; Zhang, L. The Clinical Importance of Campylobacter concisus and Other Human Hosted Campylobacter Species. Front. Cell. Infect. Microbiol. 2018, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Iraola, G. Pathogenomics of Emerging Campylobacter Species. Clin. Microbiol. Rev. 2019, 32, e00072-18. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.R.; Hoekstra, R.M.; Samuel, M.; Marcus, R.; Bender, J.; Shiferaw, B.; Reddy, S.; Ahuja, S.D.; Helfrick, D.L.; Hardnett, F.; et al. Risk Factors for Sporadic Campylobacter Infection in the United States: A Case-Control Study in FoodNet Sites. Clin. Infect. Dis. 2004, 38, S285–S296. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). National Outbreak Reporting System (NORS): NORS Dashboard. Available online: https://wwwn.cdc.gov/norsdashboard/ (accessed on 3 February 2021).
- Ebel, E.D.; Williams, M.S.; Cole, D.; Travis, C.C.; Klontz, K.C.; Golden, N.J.; Hoekstra, R.M. Comparing Characteristics of Sporadic and Outbreak-Associated Foodborne Illnesses, United States, 2004–2011. Emerg. Infect. Dis. 2016, 22, 1193–1200. [Google Scholar] [CrossRef]
- Ricotta, E.E.; Palmer, A.; Wymore, K.; Clogher, P.; Oosmanally, N.; Robinson, T.; Lathrop, S.; Karr, J.; Hatch, J.; Dunn, J.; et al. Epidemiology and antimicrobial resistance of international travel-associated Campylobacter infections in the United States, 2005–2011. Am. J. Public Health 2014, 104, e108–e114. [Google Scholar] [CrossRef]
- Cody, A.J.; Maiden, M.C.; Strachan, N.J.; McCarthy, N.D. A systematic review of source attribution of human campylobacteriosis using multilocus sequence typing. Eurosurveillance 2019, 24, 1800696. [Google Scholar] [CrossRef]
- Ravel, A.; Hurst, M.; Petrica, N.; David, J.; Mutschall, S.K.; Pintar, K.; Taboada, E.N.; Pollari, F. Source attribution of human campylobacteriosis at the point of exposure by combining comparative exposure assessment and subtype comparison based on comparative genomic fingerprinting. PLoS ONE 2017, 12, e0183790. [Google Scholar] [CrossRef]
- Kittl, S.; Heckel, G.; Korczak, B.M.; Kuhnert, P. Source Attribution of Human Campylobacter Isolates by MLST and Fla-Typing and Association of Genotypes with Quinolone Resistance. PLoS ONE 2013, 8, e81796. [Google Scholar] [CrossRef]
- Mughini-Gras, L.; Pijnacker, R.; Coipan, C.; Mulder, A.C.; Fernandes Veludo, A.; de Rijk, S.; van Hoek, A.H.A.M.; Buij, R.; Muskens, G.; Koene, M.; et al. Sources and transmission routes of campylobacteriosis: A combined analysis of genome and exposure data. J. Infect. 2021, 82, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Lake, R.J.; Campbell, D.M.; Hathaway, S.C.; Ashmore, E.; Cressey, P.J.; Horn, B.J.; Pirikahu, S.; Sherwood, J.M.; Baker, M.G.; Shoemack, P.; et al. Source attributed case-control study of campylobacteriosis in New Zealand. Int. J. Infect. Dis. 2021, 103, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Mossong, J.; Mughini-Gras, L.; Penny, C.; Devaux, A.; Olinger, C.; Losch, S.; Cauchie, H.-M.; van Pelt, W.; Ragimbeau, C. Human Campylobacteriosis in Luxembourg, 2010–2013: A Case-Control Study Combined with Multilocus Sequence Typing for Source Attribution and Risk Factor Analysis. Sci. Rep. 2016, 6, 20939. [Google Scholar] [CrossRef]
- Tyson, G.H.; Tate, H.P.; Abbott, J.; Tran, T.T.; Kabera, C.; Crarey, E.; Young, S.; McDermott, P.F.; Sprague, G.; Campbell, M.; et al. Molecular Subtyping and Source Attribution of Campylobacter Isolated from Food Animals. J. Food Prot. 2016, 79, 1891–1897. [Google Scholar] [CrossRef]
- Liao, S.-J.; Marshall, J.; Hazelton, M.L.; French, N.P. Extending statistical models for source attribution of zoonotic diseases: A study of campylobacteriosis. J. R. Soc. Interface 2019, 16, 20180534. [Google Scholar] [CrossRef]
- Thépault, A.; Méric, G.; Rivoal, K.; Pascoe, B.; Mageiros, L.; Touzain, F.; Rose, V.; Béven, V.; Chemaly, M.; Sheppard, S.K. Genome-Wide Identification of Host-Segregating Epidemiological Markers for Source Attribution in Campylobacter jejuni. Appl. Environ. Microbiol. 2017, 83, e03085-16. [Google Scholar] [CrossRef]
- Thépault, A.; Rose, V.; Quesne, S.; Poezevara, T.; Béven, V.; Hirchaud, E.; Touzain, F.; Lucas, P.; Méric, G.; Mageiros, L.; et al. Ruminant and chicken: Important sources of campylobacteriosis in France despite a variation of source attribution in 2009 and 2015. Sci. Rep. 2018, 8, 9305. [Google Scholar] [CrossRef]
- Rosner, B.M.; Schielke, A.; Didelot, X.; Kops, F.; Breidenbach, J.; Willrich, N.; Gölz, G.; Alter, T.; Stingl, K.; Josenhans, C.; et al. A combined case-control and molecular source attribution study of human Campylobacter infections in Germany, 2011–2014. Sci. Rep. 2017, 7, 5139. [Google Scholar] [CrossRef]
- Hsu, C.-H.; Harrison, L.; Mukherjee, S.; Strain, E.; McDermott, P.; Zhang, Q.; Zhao, S. Core Genome Multilocus Sequence Typing for Food Animal Source Attribution of Human Campylobacter jejuni Infections. Pathogens 2020, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; Colles, F.; Richardson, J.; Cody, A.J.; Elson, R.; Lawson, A.; Brick, G.; Meldrum, R.; Little, C.L.; Owen, R.J.; et al. Host Association of Campylobacter Genotypes Transcends Geographic Variation. Appl. Environ. Microbiol. 2010, 76, 5269–5277. [Google Scholar] [CrossRef] [PubMed]
- Berthenet, E.; Thépault, A.; Chemaly, M.; Rivoal, K.; Ducournau, A.; Buissonnière, A.; Bénéjat, L.; Bessède, E.; Mégraud, F.; Sheppard, S.K.; et al. Source attribution of Campylobacter jejuni shows variable importance of chicken and ruminants reservoirs in non-invasive and invasive French clinical isolates. Sci. Rep. 2019, 9, 8098. [Google Scholar] [CrossRef] [PubMed]
- Kovac, J.; Stessl, B.; Čadež, N.; Gruntar, I.; Cimerman, M.; Stingl, K.; Lušicky, M.; Ocepek, M.; Wagner, M.; Smole Možina, S. Population structure and attribution of human clinical Campylobacter jejuni isolates from central Europe to livestock and environmental sources. Zoonoses Public Health 2018, 65, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Di Giannatale, E.; Garofolo, G.; Alessiani, A.; Di Donato, G.; Candeloro, L.; Vencia, W.; Decastelli, L.; Marotta, F. Tracing Back Clinical Campylobacter jejuni in the Northwest of Italy and Assessing Their Potential Source. Front. Microbiol. 2016, 7, 887. [Google Scholar] [CrossRef]
- Miller, W.G.; Englen, M.D.; Kathariou, S.; Wesley, I.V.; Wang, G.; Pittenger-Alley, L.; Siletz, R.M.; Muraoka, W.; Fedorka-Cray, P.J.; Mandrell, R.E. Identification of host-associated alleles by multilocus sequence typing of Campylobacter coli strains from food animals. Microbiology 2006, 152, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Jehanne, Q.; Pascoe, B.; Bénéjat, L.; Ducournau, A.; Buissonnière, A.; Mourkas, E.; Mégraud, F.; Bessède, E.; Sheppard, S.K.; Lehours, P. Genome-Wide Identification of Host-Segregating Single-Nucleotide Polymorphisms for Source Attribution of Clinical Campylobacter coli Isolates. Appl. Environ. Microbiol. 2020, 86, e01787-20. [Google Scholar] [CrossRef]
- Nohra, A.; Grinberg, A.; Midwinter, A.C.; Marshall, J.C.; Collins-Emerson, J.M.; French, N.P. Molecular Epidemiology of Campylobacter coli Strains Isolated from Different Sources in New Zealand between 2005 and 2014. Appl. Environ. Microbiol. 2016, 82, 4363–4370. [Google Scholar] [CrossRef]
- Roux, F.; Sproston, E.; Rotariu, O.; MacRae, M.; Sheppard, S.K.; Bessell, P.; Smith-Palmer, A.; Cowden, J.; Maiden, M.C.J.; Forbes, K.J.; et al. Elucidating the Aetiology of Human Campylobacter coli Infections. PLoS ONE 2013, 8, e64504. [Google Scholar] [CrossRef]
- Harrison, L.; Mukherjee, S.; Hsu, C.-H.; Young, S.; Strain, E.; Zhang, Q.; Tillman, G.E.; Morales, C.; Haro, J.; Zhao, S. Core Genome MLST for Source Attribution of Campylobacter coli. Front. Microbiol. 2021, 12, 1936. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Fast Facts about PulseNet. Available online: https://www.cdc.gov/pulsenet/about/fast-facts.html (accessed on 10 March 2021).
- Tolar, B.; Joseph, L.A.; Schroeder, M.N.; Stroika, S.; Ribot, E.M.; Hise, K.B.; Gerner-Smidt, P. An Overview of PulseNet USA Databases. Foodborne Pathog. Dis. 2019, 16, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ribot, E.M.; Freeman, M.; Hise, K.B.; Gerner-Smidt, P. PulseNet: Entering the Age of Next-Generation Sequencing. Foodborne Pathog. Dis. 2019, 16, 451–456. [Google Scholar] [CrossRef]
- Tong, S.; Ma, L.; Ronholm, J.; Hsiao, W.; Lu, X. Whole genome sequencing of Campylobacter in agri-food surveillance. Curr. Opin. Food Sci. 2021, 39, 130–139. [Google Scholar] [CrossRef]
- Denes, T.; den Bakker, H.C.; Tokman, J.I.; Guldimann, C.; Wiedmann, M. Selection and Characterization of Phage-Resistant Mutant Strains of Listeria monocytogenes Reveal Host Genes Linked to Phage Adsorption. Appl. Environ. Microbiol. 2015, 81, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S.; Fast, Q.C. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 11 March 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, B. Bbtools: A Suite of Fast, Multithreaded Bioinformatics Tools Designed for Analysis of DNA and RNA Sequence Data. Available online: https://jgi.doe.gov/data-and-tools/bbtools/ (accessed on 25 June 2021).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Gardner, S.N.; Slezak, T.; Hall, B.G. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015, 31, 2877–2878. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Dingle, K.E.; Colles, F.M.; Wareing, D.R.A.; Ure, R.; Fox, A.J.; Bolton, F.E.; Bootsma, H.J.; Willems, R.J.L.; Urwin, R.; Maiden, M.C.J. Multilocus Sequence Typing System for Campylobacter jejuni. J. Clin. Microbiol. 2001, 39, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.G.; On, S.L.W.; Wang, G.; Fontanoz, S.; Lastovica, A.J.; Mandrell, R.E. Extended Multilocus Sequence Typing System for Campylobacter coli, C. lari, C. upsaliensis, and C. helveticus. J. Clin. Microbiol. 2005, 43, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology 2012, 158, 1005–1015. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2015, 32, 929–931. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Pontén, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O.; et al. Benchmarking of Methods for Genomic Taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Davis, S.; Pettengill, J.B.; Luo, Y.; Payne, J.; Shpuntoff, A.; Rand, H.; Strain, E. CFSAN SNP Pipeline: An automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 2015, 1, e20. [Google Scholar] [CrossRef]
- Sheppard, S.K.; Didelot, X.; Jolley, K.A.; Darling, A.E.; Pascoe, B.; Meric, G.; Kelly, D.J.; Cody, A.; Colles, F.M.; Strachan, N.J.C.; et al. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol. Ecol. 2013, 22, 1051–1064. [Google Scholar] [CrossRef]
- Skarp-de Haan, C.P.A.; Culebro, A.; Schott, T.; Revez, J.; Schweda, E.K.H.; Hänninen, M.-L.; Rossi, M. Comparative genomics of unintrogressed Campylobacter coli clades 2 and 3. BMC Genom. 2014, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; Dallas, J.F.; Wilson, D.J.; Strachan, N.J.C.; McCarthy, N.D.; Jolley, K.A.; Colles, F.M.; Rotariu, O.; Ogden, I.D.; Forbes, K.J.; et al. Evolution of an Agriculture-Associated Disease Causing Campylobacter coli Clade: Evidence from National Surveillance Data in Scotland. PLoS ONE 2010, 5, e15708. [Google Scholar] [CrossRef]
- Kelley, B.R.; Ellis, J.C.; Large, A.; Schneider, L.G.; Jacobson, D.; Johnson, J.G. Whole-Genome Sequencing and Bioinformatic Analysis of Environmental, Agricultural, and Human Campylobacter jejuni Isolates From East Tennessee. Front. Microbiol. 2020, 11, 571064. [Google Scholar] [CrossRef]
- Sheppard, S.K.; Dallas, J.F.; MacRae, M.; McCarthy, N.D.; Sproston, E.L.; Gormley, F.J.; Strachan, N.J.C.; Ogden, I.D.; Maiden, M.C.J.; Forbes, K.J. Campylobacter genotypes from food animals, environmental sources and clinical disease in Scotland 2005/6. Int. J. Food Microbiol. 2009, 134, 96–103. [Google Scholar] [CrossRef]
- Sheppard, S.K.; Cheng, L.; Méric, G.; de Haan, C.P.A.; Llarena, A.-K.; Marttinen, P.; Vidal, A.; Ridley, A.; Clifton-Hadley, F.; Connor, T.R.; et al. Cryptic ecology among host generalist Campylobacter jejuni in domestic animals. Mol. Ecol. 2014, 23, 2442–2451. [Google Scholar] [CrossRef]
- USDA National Agricultural Statistics Service. 2020 State Agriculture Overview: Tennessee. Available online: https://www.nass.usda.gov/Quick_Stats/Ag_Overview/stateOverview.php?state=TENNESSEE (accessed on 25 May 2021).
- USDA National Agricultural Statistics Service. 2020 State Agriculture Overview: Pennsylvania. Available online: https://www.nass.usda.gov/Quick_Stats/Ag_Overview/stateOverview.php?state=PENNSYLVANIA (accessed on 25 May 2021).
- Käferstein, F.K. Actions to reverse the upward curve of foodborne illness. Food Control. 2003, 14, 101–109. [Google Scholar] [CrossRef]
- Majowicz, S.E.; Meyer, S.B.; Kirkpatrick, S.I.; Graham, J.L.; Shaikh, A.; Elliott, S.J.; Minaker, L.M.; Scott, S.; Laird, B. Food, health, and complexity: Towards a conceptual understanding to guide collaborative public health action. BMC Public Health 2016, 16, 487. [Google Scholar] [CrossRef]
- Pires, S.M.; Thomsen, S.T.; Nauta, M.; Poulsen, M.; Jakobsen, L.S. Food Safety Implications of Transitions Toward Sustainable Healthy Diets. Food Nutr. Bull. 2020, 41, 104S–124S. [Google Scholar] [CrossRef] [PubMed]
- Doorduyn, Y.; Van Den Brandhof, W.E.; Van Duynhoven, Y.T.H.P.; Breukink, B.J.; Wagenaar, J.A.; Van Pelt, W. Risk factors for indigenous Campylobacter jejuni and Campylobacter coli infections in The Netherlands: A case-control study. Epidemiol. Infect. 2010, 138, 1391–1404. [Google Scholar] [CrossRef] [PubMed]
- Brehony, C.; Lanigan, D.; Carroll, A.; McNamara, E. Establishment of sentinel surveillance of human clinical campylobacteriosis in Ireland. Zoonoses Public Health 2021, 68, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, S.M.; Kivistö, R.I.; Rossi, M.; Schott, T.; Kärkkäinen, U.-M.; Tuuminen, T.; Uksila, J.; Rautelin, H.; Hänninen, M.-L. Multilocus Sequence Typing (MLST) and Whole-Genome MLST of Campylobacter jejuni Isolates from Human Infections in Three Districts during a Seasonal Peak in Finland. J. Clin. Microbiol. 2014, 52, 4147–4154. [Google Scholar] [CrossRef]
- Llarena, A.-K.; Taboada, E.; Rossi, M. Whole-Genome Sequencing in Epidemiology of Campylobacter jejuni Infections. J. Clin. Microbiol. 2017, 55, 1269–1275. [Google Scholar] [CrossRef]
- Joensen, K.G.; Kuhn, K.G.; Müller, L.; Björkman, J.T.; Torpdahl, M.; Engberg, J.; Holt, H.M.; Nielsen, H.L.; Petersen, A.M.; Ethelberg, S.; et al. Whole-genome sequencing of Campylobacter jejuni isolated from Danish routine human stool samples reveals surprising degree of clustering. Clin. Microbiol. Infect. 2018, 24, 201.e205–201.e208. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Species, Clade, Subclade | Clonal Complexes | No. of Isolates (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| State | Source | |||||||||
| PA | TN | Clinical | Non-Clinical | |||||||
| Poultry | Cattle | Swine | Other | |||||||
| C. jejuni | 2132 (69.2%) | 887 (41.6%) | 1245 (58.4%) | 960 (45.0%) | 1172 (55.0%) | 735 (62.7%) | 419 (35.8%) | 15 (1.3%) | 3 (0.3%) | |
| 1 | ST-45, ST-403, ST-508, ST-179, ST-283, ST-177, ST-682, ST-41 | 251 | 84 (33.5%) | 167 (66.5%) | 169 (67.3%) | 82 (32.%) | 39 (47.6%) | 40 (48.8%) | 3 (3.7%) | 0 |
| 2 | - | 41 | 19 (46.3%) | 22 (53.7%) | 21 (51.2%) | 20 (48.8%) | 0 | 17 (85.0%) | 3 (15.0%) | 0 |
| 3 | ST-21, ST-353, ST-48, ST-206, ST-61, ST-42, ST-464, ST-607, ST-22, ST-443, ST-52, ST-257, ST-354, ST-460, ST-49, ST-658, ST-1034, ST-1287, ST-1332, ST-446, ST-692 | 1840 | 784 (42.6%) | 1056 (57.4%) | 770 (41.8%) | 1070 (58.2%) | 696 (65.0%) | 362 (33.8%) | 9 (0.8%) | 3 (0.3%) |
| C. coli | 921 (29.9%) | 529 (57.4%) | 392 (42.6%) | 98 (10.6%) | 823 (89.4%) | 504 (61.2%) | 71 (8.6%) | 239 (29.0%) | 9 (1.1%) | |
| 1 | 916 | 529 (57.8%) | 387 (42.2%) | 96 (10.5%) | 820 (89.5%) | 501 (61.1%) | 71 (8.7%) | 239 (29.1%) | 9 (1.1%) | |
| 1a | ST-828 | 897 | 516 (57.5%) | 381 (42.5%) | 93 (10.4%) | 804 (89.6%) | 486 (60.4%) | 70 (8.7%) | 239 (29.7%) | 9 (1.1%) |
| 1b | ST-1150 | 16 | 11 (68.8%) | 5 (31.3%) | 1 (6.3%) | 15 (93.8%) | 14 (93.3%) | 1 (6.7%) | 0 | 0 |
| 1c | - | 3 | 2 (66.7%) | 1 (33.3%) | 2 (66.7%) | 1 (33.3%) | 1 (100.0%) | 0 | 0 | 0 |
| 3 | - | 5 | 0 | 5 (100.0%) | 2 (40.0%) | 3 (60.0%) | 3 (100.0%) | 0 | 0 | 0 |
| C. lari | ST-21 | 13 (0.4%) | 10 (76.9%) | 3 (23.1%) | 3 (23.1%) | 10 (76.9%) | 6 (60.0%) | 4 (40.0%) | 0 | 0 |
| C. upsaliensis | - | 10 (0.3%) | 1 (10.0%) | 9 (90.0%) | 9 (90.0%) | 1 (10.0%) | 0 | 0 | 0 | 1 (100.0%) |
| C. fetus | ST-3, ST-6, ST-11 | 4 (0.1%) | 0 | 4 (100.0%) | 4 (100.0%) | 0 | 0 | 0 | 0 | 0 |
| Total | 3080 | 1427 (46.3%) | 1653 (53.7%) | 1074 (34.9%) | 2006 (65.1%) | 1244 (62.0%) | 494 (24.6%) | 254 (12.7%) | 10 (0.5%) | |
| No. Potential Clusters | ||||
|---|---|---|---|---|
| Species | Total | PA-Only | TN-Only | Related to Non-Clinical Isolates |
| C. jejuni | 42 | 1 | 33 | 8 |
| C. coli | 1 | 0 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudson, L.K.; Andershock, W.E.; Yan, R.; Golwalkar, M.; M’ikanatha, N.M.; Nachamkin, I.; Thomas, L.S.; Moore, C.; Qian, X.; Steece, R.; et al. Phylogenetic Analysis Reveals Source Attribution Patterns for Campylobacter spp. in Tennessee and Pennsylvania. Microorganisms 2021, 9, 2300. https://doi.org/10.3390/microorganisms9112300
Hudson LK, Andershock WE, Yan R, Golwalkar M, M’ikanatha NM, Nachamkin I, Thomas LS, Moore C, Qian X, Steece R, et al. Phylogenetic Analysis Reveals Source Attribution Patterns for Campylobacter spp. in Tennessee and Pennsylvania. Microorganisms. 2021; 9(11):2300. https://doi.org/10.3390/microorganisms9112300
Chicago/Turabian StyleHudson, Lauren K., William E. Andershock, Runan Yan, Mugdha Golwalkar, Nkuchia M. M’ikanatha, Irving Nachamkin, Linda S. Thomas, Christina Moore, Xiaorong Qian, Richard Steece, and et al. 2021. "Phylogenetic Analysis Reveals Source Attribution Patterns for Campylobacter spp. in Tennessee and Pennsylvania" Microorganisms 9, no. 11: 2300. https://doi.org/10.3390/microorganisms9112300
APA StyleHudson, L. K., Andershock, W. E., Yan, R., Golwalkar, M., M’ikanatha, N. M., Nachamkin, I., Thomas, L. S., Moore, C., Qian, X., Steece, R., Garman, K. N., Dunn, J. R., Kovac, J., & Denes, T. G. (2021). Phylogenetic Analysis Reveals Source Attribution Patterns for Campylobacter spp. in Tennessee and Pennsylvania. Microorganisms, 9(11), 2300. https://doi.org/10.3390/microorganisms9112300