
Pan-Genome Analysis of Effectors in Korean Strains of the Soybean Pathogen Xanthomonas citri pv. glycines

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and DNA Preparation
2.2. Genome Sequencing and Assembly
2.3. Comparative Genome Analysis
2.4. CAZyme and TALE Analysis
3. Results
3.1. Whole-Genome Sequence Analysis of Korean X. citri pv. glycines Strains
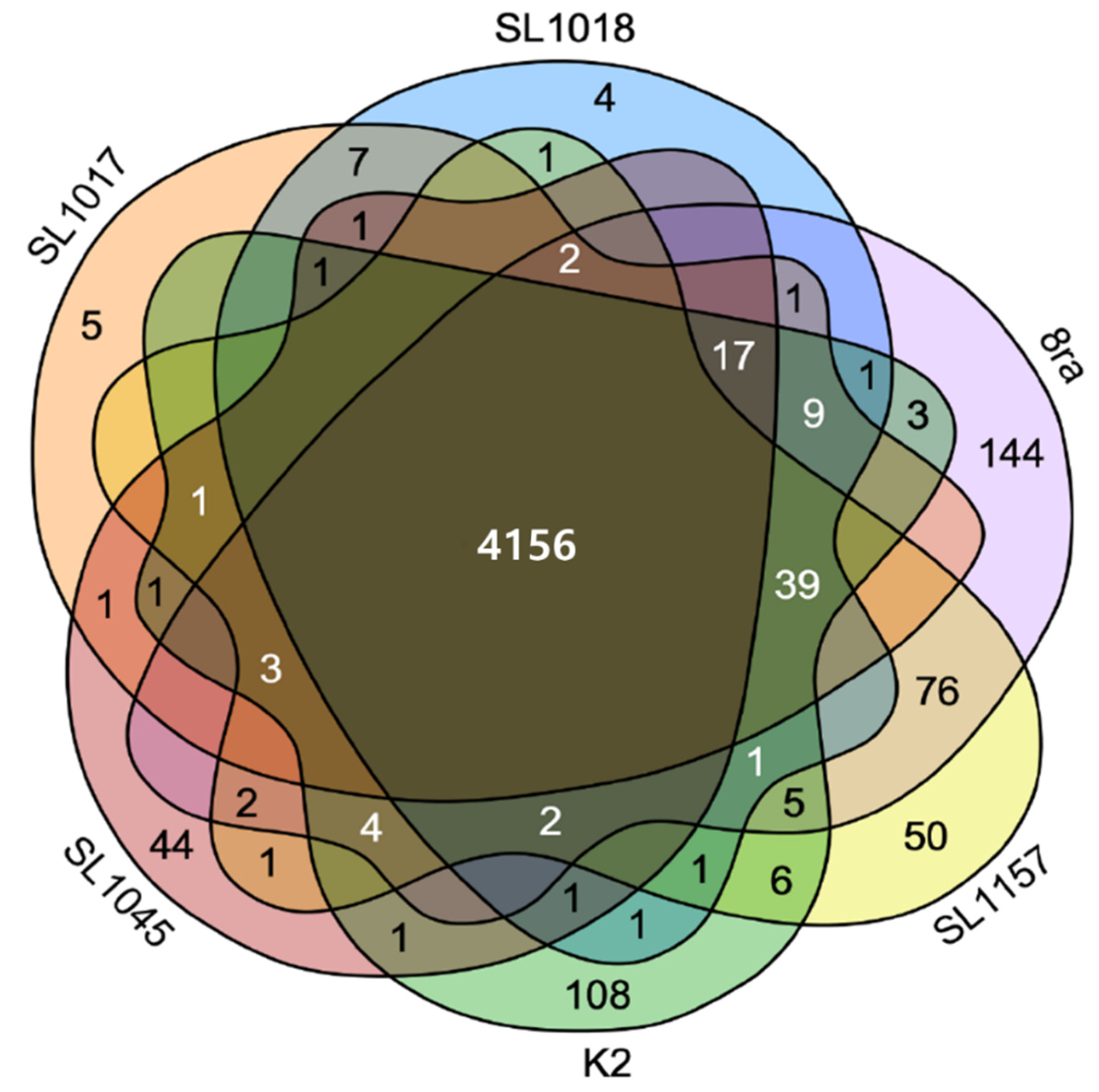
3.2. Comparison of Gene Content and Number of Korean X. citri pv. glycines Strains
3.3. Functional Gene Distribution in the Korean X. citri pv. glycines Strains and Strain 8ra
3.4. Comparison of Total Number of CAZymes Present in the Genomes of Korean X. citri pv. glycines Strains and Strain 8ra
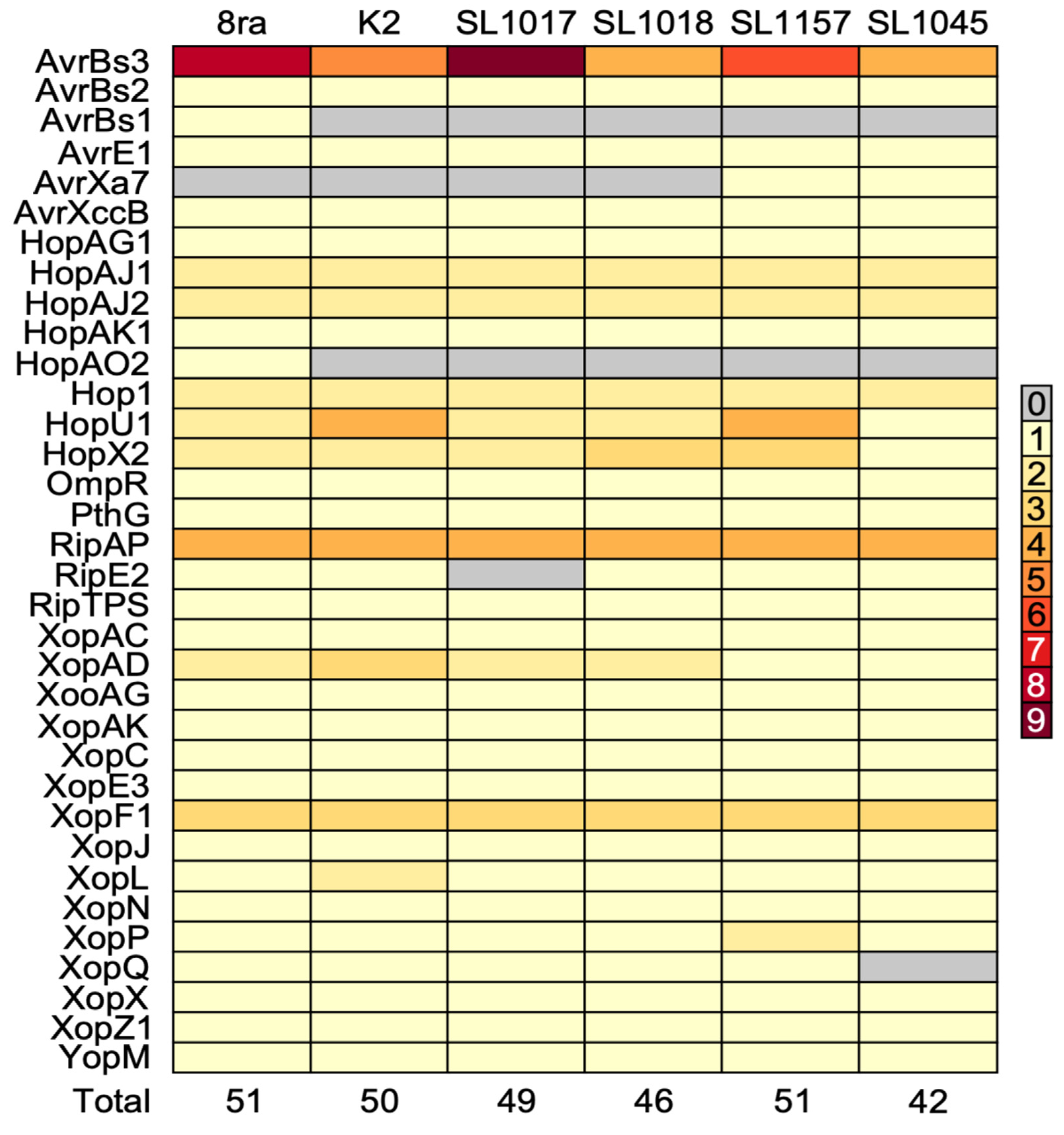
3.5. Comparative Analysis of Type III Secretion System Effector Candidate Genes
3.6. Comparative Analysis of Transcription Activator-like Effector Candidate Genes
3.7. Comparative Analysis of the Predicted Effector Binding Elements of the Transcription Activator-like Effector Candidate Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Constantin, E.C.; Cleenwerck, I.; Maes, M.; Baeyen, S.; Van Malderghem, C.; De Vos, P.; Cottyn, B. Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 2016, 65, 792–806. [Google Scholar] [CrossRef]
- Hong, S.J.; Kim, Y.K.; Jee, H.J.; Lee, B.C.; Yoon, Y.N.; Park, S.T. Selection of bactericides for controlling soybean bacterial pustule. Res. Plant Dis. 2010, 16, 266–273. (In Korean) [Google Scholar] [CrossRef]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Büttner, D.; Bonas, U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 2010, 34, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Narvel, J.M.; Jakkula, L.R.; Phillips, D.V.; Wang, T.; Lee, S.H.; Boerma, H.R. Molecular mapping of rxp conditioning reaction to bacterial pustule in soybean. J. Hered. 2001, 92, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Athinuwat, D.; Prathuangwong, S.; Cursino, L.; Burr, T. Xanthomonas axonopodis pv. glycines Soybean Cultivar Virulence Specificity Is Determined by avrBs3 Homolog avrXg1. Phytopathology 2009, 99, 996–1004. [Google Scholar] [CrossRef]
- Hwang, I.; Lim, S.M. Pathogenic variability in isolates of Xanthomonas campestris pv. glycines. Korean J. Plant Pathol. 1998, 14, 19–22. [Google Scholar]
- Park, H.J.; Han, S.W.; Oh, C.; Lee, S.; Ra, D.; Lee, S.H.; Heu, S. Avirulence gene diversity of Xanthomonas axonopodis pv. glycines isolated in Korea. J. Microbiol. Biotechnol. 2008, 18, 1500–1509. [Google Scholar]
- Kaewnum, S.; Prathuangwong, S.; Burr, T.J. Aggressiveness of Xanthomonas axonopodis pv. glycines isolates to soybean and hypersensitivity responses by other plants. Plant Pathol. 2005, 54, 409–415. [Google Scholar] [CrossRef]
- Scholze, H.; Boch, J. TAL effectors are remote controls for gene activation. Curr. Opin. Microbiol. 2011, 14, 47–53. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Gu, K.; Yang, B.; Tian, D.; Wu, L.; Wang, D.; Sreekala, C.; Yang, F.; Chu, Z.; Wang, G.L.; White, F.F.; et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature 2005, 435, 1122–1125. [Google Scholar] [CrossRef]
- Kim, J.-G.; Choi, S.; Oh, J.; Moon, J.S.; Hwang, I. Comparative analysis of three indigenous plasmids from Xanthomonas axonopodis pv. glycines. Plasmid 2006, 56, 79–87. [Google Scholar] [CrossRef]
- Oh, C.S.; Lee, S.; Heu, S. Genetic Diversity of avrBs-like Genes in Three Different Xanthomonas Species Isolated in Korea. Plant Pathol. J. 2011, 27, 26–32. [Google Scholar] [CrossRef][Green Version]
- Ji, Z.Y.; Zakria, M.; Zou, L.F.; Xiong, L.; Li, Z.; Ji, G.H.; Chen, G.Y. Genetic Diversity of Transcriptional Activator-Like Effector Genes in Chinese Isolates of Xanthomonas oryzae pv. oryzicola. Phytopathology 2014, 104, 672–682. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, B.M.; Park, Y.J.; Park, D.S.; Kang, H.W.; Kim, J.G.; Song, E.S.; Park, I.C.; Yoon, U.H.; Hahn, J.H.; Koo, B.S.; et al. The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res. 2005, 33, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; White, F.F. Diverse members of the AvrBs3/PthA family of type III effectors are major virulence determinants in bacterial blight disease of rice. Mol. Plant-Microbe Interact. 2004, 17, 1192–1200. [Google Scholar] [CrossRef]
- Chatnaparat, T.; Prathuangwong, S.; Ionescu, M.; Lindow, S.E. XagR, a LuxR Homolog, Contributes to the Virulence of Xanthomonas axonopodis pv. glycines to Soybean. Mol. Plant-Microbe Interact. 2012, 25, 1104–1117. [Google Scholar] [CrossRef]
- Darrasse, A.; Bolot, S.; Serres-Giardi, L.; Charbit, E.; Boureau, T.; Fisher-Le Saux, M.; Noel, L.D. High-quality draft genome sequences of Xanthomonas axonopodis pv. glycines strains CFBP 2526 and CFBP 7119. Genome Announc. 2013, 1, e01036-13. [Google Scholar] [CrossRef]
- Weng, S.F.; Luo, A.C.; Lin, C.J.; Tseng, T.T. High-Quality Genome Sequence of Xanthomonas axonopodis pv. glycines Strain 12609 Isolated in Taiwan. Microbiol. Resour. Announc. 2017, 5, e01695-16. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, H.; Park, H.J.; Ryu, S.; Han, S.W. Draft genome sequence of Xanthomonas axonopodis pv. glycines 8ra possessing transcription activator-like effectors used for genetic engineering. J. Biotechnol. 2014, 179, 15–16. [Google Scholar] [CrossRef]
- Carpenter, S.C.; Kladsuwan, L.; Han, S.W.; Prathuangwong, S.; Bogdanove, A.J. Complete Genome Sequences of Xanthomonas axonopodis pv. glycines Isolates from the United States and Thailand Reveal Conserved Transcription Activator-Like Effectors. Genome Biol. Evol. 2019, 11, 1380–1384. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010, 11, 119. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Jensen, L.J. eggNOG v4, 0, Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Machi, D. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2013, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.; Reschke, M.; Erkes, A.; Streubel, J.; Morgan, R.D.; Wilson, G.G.; Boch, J. AnnoTALE: Bioinformatics tools for identification, annotation, and nomenclature of TALEs from Xanthomonas genomic sequences. Sci. Rep. 2016, 6, 21077. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.; Wolf, A.; Reschke, M.; Bonas, U.; Posch, S.; Boch, J. Computational predictions provide insights into the biology of TAL effector target sites. PLoS Comput. Biol. 2013, 9, e1002962. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. Jvenn: An interactive Venn diagram viewer. BMC Bioinf. 2014, 15, 293. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Barak, J.D.; Vancheva, T.; Lefeuvre, P.; Jones, J.B.; Timilsina, S.; Minsavage, G.V.; Vallad, G.E.; Koebnik, R. Whole-Genome Sequences of Xanthomonas euvesicatoria Strains Clarify Taxonomy and Reveal a Stepwise Erosion of Type 3 Effectors. Front. Plant Sci. 2016, 7, 1805. [Google Scholar] [CrossRef]
- Potnis, N.; Krasileva, K.; Chow, V.; Almeida, N.F.; Patil, P.B.; Ryan, R.P.; Jones, J.B. Comparative genomics reveals diversity among Xanthomonads infecting tomato and pepper. BMC Genom. 2011, 12, 146. [Google Scholar] [CrossRef]
- Vieira, P.S.; Bonfim, I.M.; Araujo, E.A.; Melo, R.R.; Lima, A.R.; Fessel, M.R.; Murakami, M.T. Xyloglucan processing machinery in Xanthomonas pathogens and its role in the transcriptional activation of virulence factors. Nat. Commun. 2021, 12, 4049. [Google Scholar] [CrossRef]
- Castañeda-Ojeda, M.P.; Moreno-Pérez, A.; Ramos, C.; López-Solanilla, E. Suppression of plant immune responses by the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 type III effector tyrosine phosphatases HopAO1 and HopAO2. Front. Plant Sci. 2017, 8, 680. [Google Scholar] [CrossRef]
- Schandry, N.; de Lange, O.; Prior, P.; Lahaye, T. TALE-Like Effectors Are an Ancestral Feature of the Ralstonia solanacearum Species Complex and Converge in DNA Targeting Specificity. Front. Plant Sci. 2016, 7, 1225. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.M.; Li, Y.R.; Zou, L.F.; Chen, G.Y. Gene-for-gene relationships between rice and diverse avrBs3/pthA avirulence genes in Xanthomonas oryzae pv. oryzae. Plant Pathol. 2007, 56, 26–34. [Google Scholar] [CrossRef]
- Gochez, A.M.; Huguet-Tapia, J.C.; Minsavage, G.V.; Shantaraj, D.; Jalan, N.; Strauß, A.; Potnis, N. Pacbio sequencing of copper-tolerant Xanthomonas citri reveals presence of a chimeric plasmid structure and provides insights into reassortment and shuffling of transcription activator-like effectors among X. citri strains. BMC Genom. 2018, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Hummel, A.W.; Wilkins, K.E.; Wang, L.; Cernadas, R.A.; Bogdanove, A.J. A transcription activator-like effector from Xanthomonas oryzae pv. oryzicola elicits dose-dependent resistance in rice. Mol. Plant Pathol. 2017, 18, 55–66. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | No. of Contigs | Depth of Coverage (x) | Genome Size (bp) | % GC | No. of CDSs | No. of Unique CDSs | NCBI Accession Number |
|---|---|---|---|---|---|---|---|
| 8ra | 2 | 178 | 5,426,838 | 64.6 | 4597 | 144 | CP041781, CP041782 |
| K2 | 2 | 108 | 5,322,598 | 64.6 | 4519 | 108 | CP041966, CP041967 |
| SL1017 | 3 | 198 | 5,197,177 | 64.7 | 4341 | 5 | VMHQ00000000 |
| SL1018 | 2 | 163 | 5,162,305 | 64.7 | 4314 | 5 | CP041961, CP041962 |
| SL1157 | 2 | 175 | 5,292,354 | 64.6 | 4490 | 4 | CP041963, CP041964 |
| SL1045 | 5 | 123 | 5,169,163 | 64.7 | 4332 | 44 | VMHR00000000 |
| Strain Name | AnnoTALE Name | No. of Repeats | RVDs (the 12th and 13th Amino Acid Residues of Each Repeat Region) |
|---|---|---|---|
| 8ra | TalGN3 8ra | 22.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG-HD-HD-NG |
| TalGR3 8ra | 18.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| TalGO3 8ra | 17.5 | NI-HD-NS-NI-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG | |
| TalGQ3 8ra | 17.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NI-NG-NS-HD-NS-HD-NI-NG-NG | |
| TalGP3 8ra | 14.5 | NI-NI-NG-HD-NG-NG-HD-NI-NS-HD-NS-HD-NI-NG-NG | |
| TalGM3 8ra | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| TalGM4 8ra | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| K2 | TalGN3 K2 | 22.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG-HD-HD-NG |
| TalGR3 K2 | 18.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| TalGO3 K2 | 17.5 | NI-HD-NS-NI-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG | |
| TalGQ3 K2 | 17.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NI-NG-NS-HD-NS-HD-NI-NG-NG | |
| TalGP3 K2 | 14.5 | NI-NI-NG-HD-NG-NG-HD-NI-NS-HD-NS-HD-NI-NG-NG | |
| SL1017 | TalGN3 SL1017 | 22.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NS-HD-HD-NG |
| TalGO3 SL1017 | 17.5 | NI-HD-NS-NI-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG | |
| TalHQ1 SL1017 | 16.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NG-NS-HD-NI-HD-NI-NG-NG | |
| TalGP3 SL1017 | 14.5 | NI-NI-NG-HD-NG-NG-HD-NI-NS-HD-NS-HD-NI-NG-NG | |
| TalGR4 SL1017 | 12.5 | NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| TalGM3 SL1017 | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| TalGR3 SL1017 | 18.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| TalGO4 SL1017 | 17.5 | NI-HD-NS-NI-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG | |
| SL1018 | TalGN3 SL1018 | 34.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NS-HD-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG |
| TalGQ3 SL1018 | 17.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NI-NG-NS-HD-NI-HD-NI-NG-NG | |
| TalGM3 SL1018 | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| TalGM4 SL1018 | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| SL1157 | TalGN3 SL1157 | 22.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NS-HD-HD-NG |
| TalGO3 SL1157 | 17.5 | NI-HD-NS-NI-HD-NS-NS-NS-NS-HD-HD-HD-NG-NI-NS-HD-HD-NG | |
| TalGQ3 SL1157 | 17.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NI-NG-NS-HD-NS-HD-NI-NG-NG | |
| TalGP3 SL1157 | 14.5 | NI-NI-NG-HD-NG-NG-HD-NI-NI-HD-NS-HD-NI-NG-NG | |
| TalGM3 SL1157 | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG | |
| TalGR3 SL1157 | 18.5 | NI-HD-NS-NS-NI-NS-NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| SL1045 | TalGM3 SL1045 | 19.5 | NI-NS-HD-NG-HD-NG-NG-HD-NI-NG-NI-NI-HD-NI-HD-NI-HD-NI-NG-NG |
| TalGR3 SL1045 | 18.5 | NI-HD-NS-NS-NI-NI-NS-HD-NS-NI-NG-NI-NI-HD-NI-HD-NI-NG-NG | |
| TalGQ3 SL1045 | 17.5 | NI-NS-HD-NI-HD-NG-HD-NG-HD-NI-NG-NS-HD-NS-HD-NI-NG-NG | |
| TalGP3 SL1045 | 14.5 | NI-NI-NG-HD-NG-NG-HD-NI-NS-HD-NS-HD-NI-NG-NG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, I.-J.; Kim, K.S.; Beattie, G.A.; Yang, J.-W.; Sohn, K.H.; Heu, S.; Hwang, I. Pan-Genome Analysis of Effectors in Korean Strains of the Soybean Pathogen Xanthomonas citri pv. glycines. Microorganisms 2021, 9, 2065. https://doi.org/10.3390/microorganisms9102065
Kang I-J, Kim KS, Beattie GA, Yang J-W, Sohn KH, Heu S, Hwang I. Pan-Genome Analysis of Effectors in Korean Strains of the Soybean Pathogen Xanthomonas citri pv. glycines. Microorganisms. 2021; 9(10):2065. https://doi.org/10.3390/microorganisms9102065
Chicago/Turabian StyleKang, In-Jeong, Kyung Seok Kim, Gwyn A. Beattie, Jung-Wook Yang, Kee Hoon Sohn, Sunggi Heu, and Ingyu Hwang. 2021. "Pan-Genome Analysis of Effectors in Korean Strains of the Soybean Pathogen Xanthomonas citri pv. glycines" Microorganisms 9, no. 10: 2065. https://doi.org/10.3390/microorganisms9102065
APA StyleKang, I.-J., Kim, K. S., Beattie, G. A., Yang, J.-W., Sohn, K. H., Heu, S., & Hwang, I. (2021). Pan-Genome Analysis of Effectors in Korean Strains of the Soybean Pathogen Xanthomonas citri pv. glycines. Microorganisms, 9(10), 2065. https://doi.org/10.3390/microorganisms9102065