
Learning from Yeast about Mitochondrial Carriers


Abstract
1. Introduction
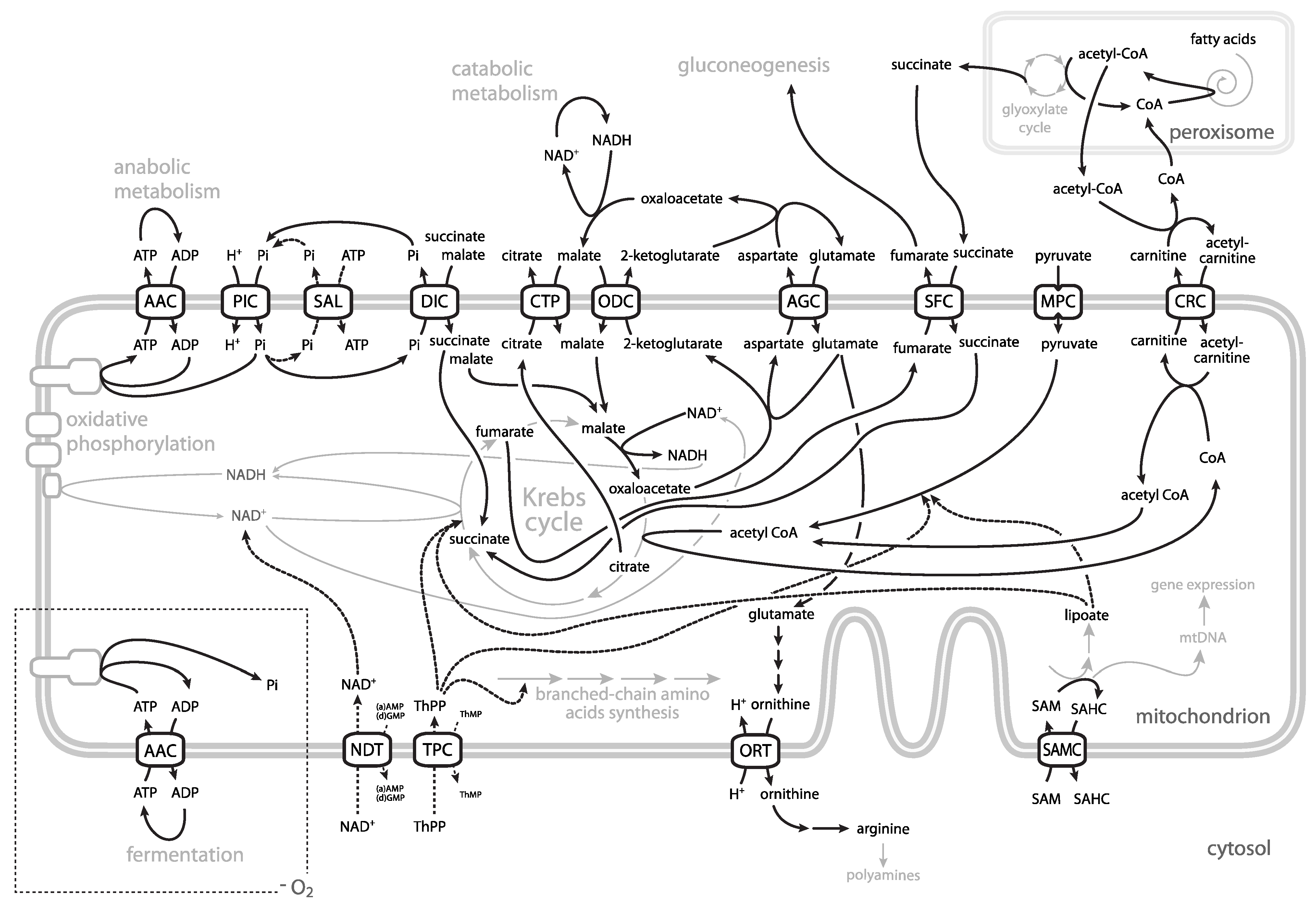
2. Mitochondrial Metabolism and Genetics
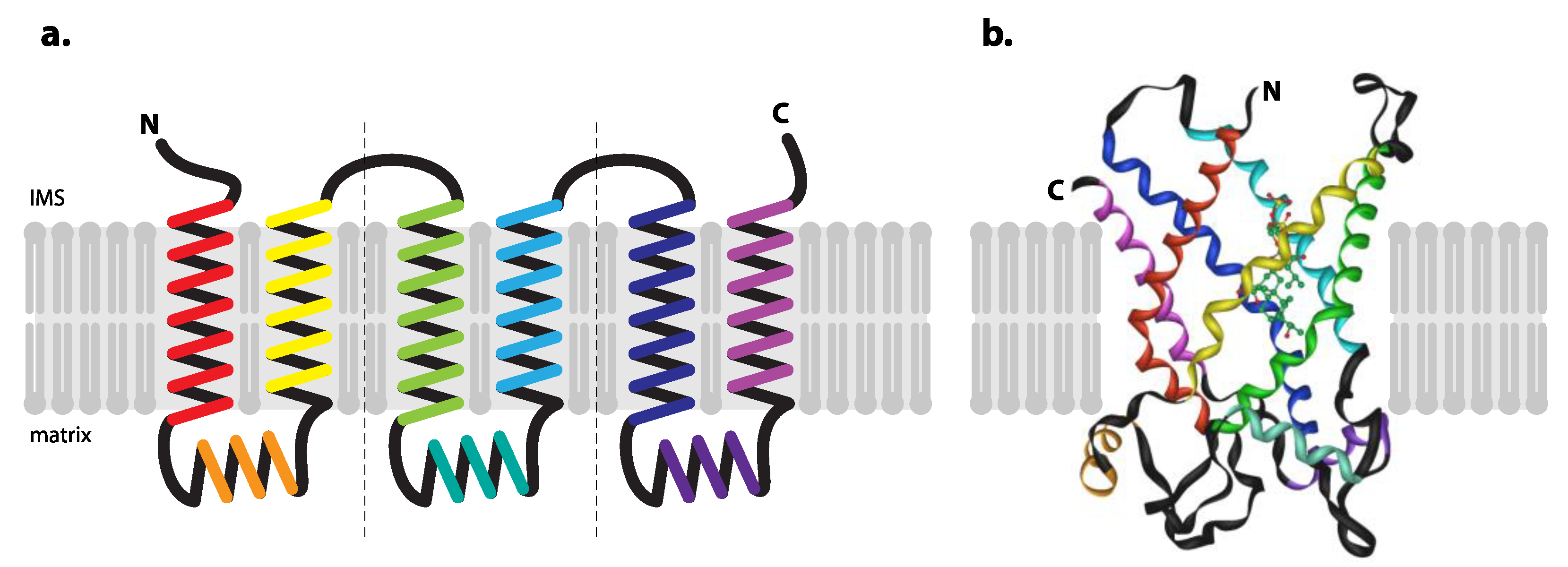
3. ADP/ATP Carrier
3.1. Human ADP/ATP Carrier
3.2. Autosomal Dominant Progressive External Ophthalmoplegia (adPEO)
3.3. Autosomal Recessive mtDNA Depletion Syndrome 12b
3.4. Autosomal Dominant Mitochondrial DNA Depletion Syndrome 12a
3.5. Drug Screening
4. ATP-Mg/Pi Carrier
Human Diseases Associated with ATP-Mg/Pi Carrier
5. Phosphate Carrier
Human Diseases Associated with Phosphate Carrier
6. Pyruvate Carrier
Human Diseases Associated with Pyruvate Carrier
7. Aspartate-Glutamate, Oxodicarboxylate and Dicarboxylate Carrier
7.1. Human Diseases Associated with Aspartate-Glutamate Carrier
7.2. Human Diseases Associated with Oxodicarboxylate Carrier
7.3. Human Diseases Associated with Dicarboxylate Carrier
8. Citrate Carrier
Human Diseases Associated with Citrate Carrier
9. NAD+ Carrier
10. Thiamine Pyrophosphate Carrier
Human Diseases Associated with Thiamine Pyrophosphate Carrier
11. S-Adenosylmethionine Carrier
Human Diseases Associated with S-Adenosylmethionine Carrier
12. Ornithine/(Citrulline) Carrier
Human Diseases Associated with Ornithine/Citrulline Carrier
13. Carnitine Carrier
Human Diseases Associated with Carnitine Carrier
14. Other Yeast Mitochondrial Carriers
15. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAC | ADP/ATP carrier |
| APC | ATP-Mg/Pi carrier |
| adPEO | autosomal dominant progressive external ophthalmoplegia |
| AGC | aspartate-glutamate carrier |
| CIC | citrate carrier |
| DIC | dicarboxylate carrier |
| HHH | hyperornithinaemia-hyperammonaemia-homocitrullinuria |
| mtDNA | mitochondrial DNA |
| ODC | oxodicarboxylate carrier |
| ORC (ORT) | ornithine carrier |
| MCF | mitochondrial carrier family |
| MPC | mitochondrial pyruvate carrier |
| SAM | S-adenosylmethionine |
| ThPP | thiamine pyrophosphate |
References
- Botstein, D.; Chervitz, S.A.; Cherry, J.M. Yeast as a model organism. Science 1997, 277, 1259–1260. [Google Scholar] [CrossRef]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, E.; Sharifpoor, S.; Baryshnikova, A.; Costanzo, M.; Myers, C.L.; Andrews, B.J.; Boone, C. Synthetic genetic array analysis for global mapping of genetic networks in yeast. Methods Mol. Biol. 2014, 1205, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.M.; Jaspersen, S.L. Manipulating the yeast genome: Deletion, mutation, and tagging by PCR. Methods Mol. Biol. 2014, 1205, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, R. Targeting, disruption, replacement, and allele rescue: Integrative DNA transformation in yeast. Methods Enzymol. 1991, 194, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Faye, G.; Fukuhara, H.; Grandchamp, C.; Lazowska, J.; Michel, F.; Casey, J.; Getz, G.S.; Locker, J.; Rabinowitz, M.; Bolotin-Fukuhara, M.; et al. Mitochondrial nucleic acids in the petite colonie mutants: Deletions and repetition of genes. Biochimie 1973, 55, 779–792. [Google Scholar] [CrossRef]
- Heude, M.; Fukuhara, H.; Moustacchi, E. Spontaneous and induced rho mutants of Saccharomyces cerevisiae: Patterns of loss of mitochondrial genetic markers. J. Bacteriol. 1979, 139, 460–467. [Google Scholar] [CrossRef]
- Solieri, L. Mitochondrial inheritance in budding yeasts: Towards an integrated understanding. Trends Microbiol. 2010, 18, 521–530. [Google Scholar] [CrossRef]
- Bullerwell, C.E.; Lang, B.F. Fungal evolution: The case of the vanishing mitochondrion. Curr. Opin. Microbiol. 2005, 8, 362–369. [Google Scholar] [CrossRef]
- Lill, R.; Diekert, K.; Kaut, A.; Lange, H.; Pelzer, W.; Prohl, C.; Kispal, G. The essential role of mitochondria in the biogenesis of cellular iron-sulfur proteins. Biol. Chem. 1999, 380, 1157–1166. [Google Scholar] [CrossRef]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, A.A.; Stier, T.J. Anaerobic nutrition of Saccharomyces cerevisiae. I. Ergosterol requirement for growth in a defined medium. J. Cell Comp. Physiol. 1953, 41, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Parks, L.W. Metabolism of sterols in yeast. CRC Crit. Rev. Microbiol. 1978, 6, 301–341. [Google Scholar] [CrossRef] [PubMed]
- Waldbauer, J.R.; Newman, D.K.; Summons, R.E. Microaerobic steroid biosynthesis and the molecular fossil record of Archean life. Proc. Natl. Acad. Sci. USA 2011, 108, 13409–13414. [Google Scholar] [CrossRef] [PubMed]
- Piškur, J.; Rozpedowska, E.; Poláková, S.; Merico, A.; Compagno, C. How did Saccharomyces evolve to become a good brewer? Trends Genet. 2006, 22, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T.; Yde Steensma, H.; Van Dijken, J.P. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 1996, 12, 1607–1633. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Rozpedowska, E.; Hellborg, L.; Ishchuk, O.P.; Orhan, F.; Galafassi, S.; Merico, A.; Woolfit, M.; Compagno, C.; Piskur, J. Parallel evolution of the make-accumulate-consume strategy in Saccharomyces and Dekkera yeasts. Nat. Commun. 2011, 2, 302. [Google Scholar] [CrossRef]
- Hagman, A.; Sall, T.; Compagno, C.; Piskur, J. Yeast “make-accumulate-consume” life strategy evolved as a multi-step process that predates the whole genome duplication. PLoS ONE 2013, 8, e68734. [Google Scholar] [CrossRef]
- Martin, W.F.; Tielens, A.G.M.; Mentel, M. Mitochondria and Anaerobic Energy Metabolism in Eukaryotes: Biochemistry and Evolution; De Gruyter: Dusseldorf, Germany, 2021. [Google Scholar]
- Kovac, L.; Lachowicz, T.M.; Slonimski, P.P. Biochemical genetics of oxidative phosphorylation. Science 1967, 158, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Kolarov, J.; Subik, J.; Kovac, L. Oxidative phosphorylation in yeast. IX. Modification of the mitochondrial adenine nucleotide translocation system in the oxidative phosphorylation-deficient mutant op 1. Biochim. Biophys. Acta 1972, 267, 465–478. [Google Scholar] [CrossRef]
- Kolarov, J.; Klingenberg, M. The adenine nucleotide translocator in genetically and physiologically modified yeast mitochondria. FEBS Lett. 1974, 45, 320–323. [Google Scholar] [CrossRef]
- Pfaff, E.; Klingenberg, M.; Heldt, H.W. Unspecific permeation and specific exchange of adenine nucleotides in liver mitochondria. Biochim. Biophys. Acta 1965, 104, 312–315. [Google Scholar] [CrossRef]
- Duee, E.D.; Vignais, P.V. Exchange between extra- and intramitochondrial adenine nucleotides. Biochim. Biophys. Acta 1965, 107, 184–188. [Google Scholar] [CrossRef]
- O’Malley, K.; Pratt, P.; Robertson, J.; Lilly, M.; Douglas, M.G. Selection of the nuclear gene for the mitochondrial adenine nucleotide translocator by genetic complementation of the op1 mutation in yeast. J. Biol. Chem. 1982, 257, 2097–2103. [Google Scholar] [CrossRef]
- Aquila, H.; Misra, D.; Eulitz, M.; Klingenberg, M. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe Seylers Z. Physiol. Chem. 1982, 363, 345–349. [Google Scholar]
- Lawson, J.E.; Douglas, M.G. Separate genes encode functionally equivalent ADP/ATP carrier proteins in Saccharomyces cerevisiae. Isolation and analysis of AAC2. J. Biol. Chem. 1988, 263, 14812–14818. [Google Scholar] [CrossRef]
- Kolarov, J.; Kolarova, N.; Nelson, N. A third ADP/ATP translocator gene in yeast. J. Biol. Chem. 1990, 265, 12711–12716. [Google Scholar] [CrossRef]
- Gbelska, Y.; Subik, J.; Svoboda, A.; Goffeau, A.; Kovac, L. Intramitochondrial ATP and cell functions: Yeast cells depleted of intramitochondrial ATP lose the ability to grow and multiply. Eur. J. Biochem. 1983, 130, 281–286. [Google Scholar] [CrossRef]
- Kováč, L.; Kolarov, J.; Šubík, J. Genetic determination of the mitochondrial adenine nucleotide translocation system and its role in the eukaryotic cell. Mol. Cell Biochem. 1977, 14, 11–14. [Google Scholar] [CrossRef]
- Subik, J.; Kolarov, J.; Kovac, L. Obligatory requirement of intramitochondrial ATP for normal functioning of the eucaryotic cell. Biochem. Biophys. Res. Commun. 1972, 49, 192–198. [Google Scholar] [CrossRef]
- Drgon, T.; Sabova, L.; Nelson, N.; Kolarov, J. ADP/ATP translocator is essential only for anaerobic growth of yeast Saccharomyces cerevisiae. FEBS Lett. 1991, 289, 159–162. [Google Scholar] [CrossRef]
- Traba, J.; Satrustegui, J.; del Arco, A. Adenine nucleotide transporters in organelles: Novel genes and functions. Cell Mol. Life Sci. 2011, 68, 1183–1206. [Google Scholar] [CrossRef] [PubMed]
- Traba, J.; Satrustegui, J.; del Arco, A. Transport of adenine nucleotides in the mitochondria of Saccharomyces cerevisiae: Interactions between the ADP/ATP carriers and the ATP-Mg/Pi carrier. Mitochondrion 2009, 9, 79–85. [Google Scholar] [CrossRef]
- Betina, S.; Gavurnikova, G.; Haviernik, P.; Sabova, L.; Kolarov, J. Expression of the AAC2 gene encoding the major mitochondrial ADP/ATP carrier in Saccharomyces cerevisiae is controlled at the transcriptional level by oxygen, heme and HAP2 factor. Eur. J. Biochem. 1995, 229, 651–657. [Google Scholar] [CrossRef]
- Gavurnikova, G.; Sabova, L.; Kissova, I.; Haviernik, P.; Kolarov, J. Transcription of the AAC1 gene encoding an isoform of mitochondrial ADP/ATP carrier in Saccharomyces cerevisiae is regulated by oxygen in a heme-independent manner. Eur. J. Biochem. 1996, 239, 759–763. [Google Scholar] [CrossRef]
- Šabová, Ľ.; Zeman, I.; Supek, F.; Kolarov, J. Transcriptional control of AAC3 gene encoding mitochondrial ADP/ATP translocator in Saccharomyces cerevisiae by oxygen, heme and ROX1 factor. Eur. J. Biochem. 1993, 213, 547–553. [Google Scholar] [CrossRef]
- Runswick, M.J.; Powell, S.J.; Nyren, P.; Walker, J.E. Sequence of the bovine mitochondrial phosphate carrier protein: Structural relationship to ADP/ATP translocase and the brown fat mitochondria uncoupling protein. EMBO J. 1987, 6, 1367–1373. [Google Scholar] [CrossRef]
- Capobianco, L.; Brandolin, G.; Palmieri, F. Transmembrane topography of the mitochondrial phosphate carrier explored by peptide-specific antibodies and enzymatic digestion. Biochemistry 1991, 30, 4963–4969. [Google Scholar] [CrossRef]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Zara, V. Transmembrane topology, genes, and biogenesis of the mitochondrial phosphate and oxoglutarate carriers. J. Bioenerg. Biomembr. 1993, 25, 493–501. [Google Scholar] [CrossRef]
- Bisaccia, F.; Capobianco, L.; Brandolin, G.; Palmieri, F. Transmembrane topography of the mitochondrial oxoglutarate carrier assessed by peptide-specific antibodies and enzymatic cleavage. Biochemistry 1994, 33, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, L.; Bisaccia, F.; Michel, A.; Sluse, F.E.; Palmieri, F. The N- and C-termini of the tricarboxylate carrier are exposed to the cytoplasmic side of the inner mitochondrial membrane. FEBS Lett. 1995, 357, 297–300. [Google Scholar] [CrossRef]
- Nury, H.; Dahout-Gonzalez, C.; Trezeguet, V.; Lauquin, G.J.; Brandolin, G.; Pebay-Peyroula, E. Relations between structure and function of the mitochondrial ADP/ATP carrier. Annu. Rev. Biochem. 2006, 75, 713–741. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends. Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Walker, J.E. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett. 1982, 144, 250–254. [Google Scholar] [CrossRef]
- Kunji, E.R.; Harding, M. Projection structure of the atractyloside-inhibited mitochondrial ADP/ATP carrier of Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 36985–36988. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trezeguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Dolce, V.; Scarcia, P.; Iacopetta, D.; Palmieri, F. A fourth ADP/ATP carrier isoform in man: Identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett. 2005, 579, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Biol. Chem. 1992, 267, 14592–14597. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef]
- Doerner, A.; Pauschinger, M.; Badorff, A.; Noutsias, M.; Giessen, S.; Schulze, K.; Bilger, J.; Rauch, U.; Schultheiss, H.P. Tissue-specific transcription pattern of the adenine nucleotide translocase isoforms in humans. FEBS Lett. 1997, 414, 258–262. [Google Scholar] [CrossRef]
- Hatanaka, T.; Takemoto, Y.; Hashimoto, M.; Majima, E.; Shinohara, Y.; Terada, H. Significant expression of functional human type 1 mitochondrial ADP/ATP carrier in yeast mitochondria. Biol. Pharm. Bull. 2001, 24, 595–599. [Google Scholar] [CrossRef]
- Hamazaki, T.; Leung, W.Y.; Cain, B.D.; Ostrov, D.A.; Thorsness, P.E.; Terada, N. Functional expression of human adenine nucleotide translocase 4 in Saccharomyces cerevisiae. PLoS ONE 2011, 6, e19250. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, D.; Matsuyama, H.; Hamazaki, T.; Shiratsuchi, T.; Terada, N.; Hook, D.J.; Walters, M.A.; Georg, G.I.; Hawkinson, J.E. Human Adenine Nucleotide Translocase (ANT) Modulators Identified by High-Throughput Screening of Transgenic Yeast. J. Biomol. Screen. 2016, 21, 381–390. [Google Scholar] [CrossRef][Green Version]
- Swan, G.E. The pharmacology of halogenated salicylanilides and their anthelmintic use in animals. J. S. Afr. Vet. Assoc. 1999, 70, 61–70. [Google Scholar] [CrossRef]
- Zhao, X.; Spanjaard, R.A. The apoptotic action of the retinoid CD437/AHPN: Diverse effects, common basis. J. Biomed. Sci. 2003, 10, 44–49. [Google Scholar] [CrossRef]
- Merarchi, M.; Jung, Y.Y.; Fan, L.; Sethi, G.; Ahn, K.S. A Brief Overview of the Antitumoral Actions of Leelamine. Biomedicines 2019, 7, 53. [Google Scholar] [CrossRef]
- Jaiquel Baron, S.; King, M.S.; Kunji, E.R.S.; Schirris, T.J.J. Characterization of drug-induced human mitochondrial ADP/ATP carrier inhibition. Theranostics 2021, 11, 5077–5091. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mishra, T.; Thacker, G.; Mishra, M.; Narender, T.; Trivedi, A.K. Chebulinic acid inhibits MDA-MB-231 breast cancer metastasis and promotes cell death through down regulation of SOD1 and induction of autophagy. Cell Biol. Int. 2020, 44, 2553–2569. [Google Scholar] [CrossRef] [PubMed]
- Zeelen, J.; van Straaten, M.; Verdi, J.; Hempelmann, A.; Hashemi, H.; Perez, K.; Jeffrey, P.D.; Halg, S.; Wiedemar, N.; Maser, P.; et al. Structure of trypanosome coat protein VSGsur and function in suramin resistance. Nat. Microbiol. 2021, 6, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A. Suramin: A novel antineoplastic agent with multiple potential mechanisms of action. Cancer Res. 1993, 53, 2239–2248. [Google Scholar] [PubMed]
- Villalona-Calero, M.A.; Otterson, G.A.; Wientjes, M.G.; Weber, F.; Bekaii-Saab, T.; Young, D.; Murgo, A.J.; Jensen, R.; Yeh, T.K.; Wei, Y.; et al. Noncytotoxic suramin as a chemosensitizer in patients with advanced non-small-cell lung cancer: A phase II study. Ann. Oncol. 2008, 19, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Kaukonen, J. Diseases caused by nuclear genes affecting mtDNA stability. Am. J. Med. Genet. 2001, 106, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttala, A.; Zeviani, M.; Comi, G.P.; Keranen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef]
- Simoncini, C.; Siciliano, G.; Tognoni, G.; Mancuso, M. Mitochondrial ANT-1 related adPEO leading to cognitive impairment: Is there a link? Acta Myol. 2017, 36, 25–27. [Google Scholar]
- Bauer, M.K.; Schubert, A.; Rocks, O.; Grimm, S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 1999, 147, 1493–1502. [Google Scholar] [CrossRef]
- Chen, X.J. Induction of an unregulated channel by mutations in adenine nucleotide translocase suggests an explanation for human ophthalmoplegia. Hum. Mol. Genet. 2002, 11, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- De Marcos Lousa, C.; Trezeguet, V.; Dianoux, A.C.; Brandolin, G.; Lauquin, G.J. The human mitochondrial ADP/ATP carriers: Kinetic properties and biogenesis of wild-type and mutant proteins in the yeast S. cerevisiae. Biochemistry 2002, 41, 14412–14420. [Google Scholar] [CrossRef]
- Napoli, L.; Bordoni, A.; Zeviani, M.; Hadjigeorgiou, G.M.; Sciacco, M.; Tiranti, V.; Terentiou, A.; Moggio, M.; Papadimitriou, A.; Scarlato, G.; et al. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology 2001, 57, 2295–2298. [Google Scholar] [CrossRef]
- Siciliano, G.; Tessa, A.; Petrini, S.; Mancuso, M.; Bruno, C.; Grieco, G.S.; Malandrini, A.; DeFlorio, L.; Martini, B.; Federico, A.; et al. Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul. Disord. 2003, 13, 162–165. [Google Scholar] [CrossRef]
- Komaki, H.; Fukazawa, T.; Houzen, H.; Yoshida, K.; Nonaka, I.; Goto, Y. A novel D104G mutation in the adenine nucleotide translocator 1 gene in autosomal dominant progressive external ophthalmoplegia patients with mitochondrial DNA with multiple deletions. Ann. Neurol. 2002, 51, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Hudson, G.; Muller, T.; Taylor, R.W.; Chinnery, P.F.; Zierz, S. A novel ANT1 gene mutation with probable germline mosaicism in autosomal dominant progressive external ophthalmoplegia. Neuromuscul. Disord. 2005, 15, 311–315. [Google Scholar] [CrossRef]
- Fontanesi, F.; Palmieri, L.; Scarcia, P.; Lodi, T.; Donnini, C.; Limongelli, A.; Tiranti, V.; Zeviani, M.; Ferrero, I.; Viola, A.M. Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum. Mol. Genet. 2004, 13, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Lodi, T.; Bove, C.; Fontanesi, F.; Viola, A.M.; Ferrero, I. Mutation D104G in ANT1 gene: Complementation study in Saccharomyces cerevisiae as a model system. Biochem. Biophys. Res. Commun. 2006, 341, 810–815. [Google Scholar] [CrossRef]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Salinas, K.; Zuo, X.; Kucejova, B.; Chen, X.J. Dominant membrane uncoupling by mutant adenine nucleotide translocase in mitochondrial diseases. Hum. Mol. Genet. 2008, 17, 4036–4044. [Google Scholar] [CrossRef]
- Wang, X.; Zuo, X.; Kucejova, B.; Chen, X.J. Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat. Cell Biol. 2008, 10, 1090–1097. [Google Scholar] [CrossRef]
- Esposito, L.A.; Melov, S.; Panov, A.; Cottrell, B.A.; Wallace, D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl. Acad. Sci. USA 1999, 96, 4820–4825. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Chen, X.J. Misfolding of mutant adenine nucleotide translocase in yeast supports a novel mechanism of Ant1-induced muscle diseases. Mol. Biol. Cell 2015, 26, 1985–1994. [Google Scholar] [CrossRef]
- Ogunbona, O.B.; Baile, M.G.; Claypool, S.M. Cardiomyopathy-associated mutation in the ADP/ATP carrier reveals translation-dependent regulation of cytochrome c oxidase activity. Mol. Biol. Cell 2018, 29, 1449–1464. [Google Scholar] [CrossRef]
- Korver-Keularts, I.M.; de Visser, M.; Bakker, H.D.; Wanders, R.J.; Vansenne, F.; Scholte, H.R.; Dorland, L.; Nicolaes, G.A.; Spaapen, L.M.; Smeets, H.J.; et al. Two Novel Mutations in the SLC25A4 Gene in a Patient with Mitochondrial Myopathy. JIMD Rep. 2015, 22, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Heidkamper, D.; Muller, V.; Nelson, D.R.; Klingenberg, M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations on transport and the four ATP versus ADP exchange modes. Biochemistry 1996, 35, 16144–16152. [Google Scholar] [CrossRef]
- Nelson, D.R.; Lawson, J.E.; Klingenberg, M.; Douglas, M.G. Site-directed mutagenesis of the yeast mitochondrial ADP/ATP translocator. Six arginines and one lysine are essential. J. Mol. Biol. 1993, 230, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- von Renesse, A.; Morales-Gonzalez, S.; Gill, E.; Salomons, G.S.; Stenzel, W.; Schuelke, M. Muscle Weakness, Cardiomyopathy, and L-2-Hydroxyglutaric Aciduria Associated with a Novel Recessive SLC25A4 Mutation. JIMD Rep. 2019, 43, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 860–876. [Google Scholar] [CrossRef]
- Kunji, E.R.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G.; et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta 2016, 1863, 2379–2393. [Google Scholar] [CrossRef]
- Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T. Dominance of yeast aac2(R96H) and aac2(R252G) mutations, equivalent to pathological mutations in ant1, is due to gain of function. Biochem. Biophys. Res. Commun. 2017, 493, 909–913. [Google Scholar] [CrossRef]
- King, M.S.; Thompson, K.; Hopton, S.; He, L.; Kunji, E.R.S.; Taylor, R.W.; Ortiz-Gonzalez, X.R. Expanding the phenotype of de novo SLC25A4-linked mitochondrial disease to include mild myopathy. Neurol. Genet. 2018, 4, e256. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef]
- Muller, V.; Heidkamper, D.; Nelson, D.R.; Klingenberg, M. Mutagenesis of some positive and negative residues occurring in repeat triad residues in the ADP/ATP carrier from yeast. Biochemistry 1997, 36, 16008–16018. [Google Scholar] [CrossRef]
- Nelson, D.R.; Felix, C.M.; Swanson, J.M. Highly conserved charge-pair networks in the mitochondrial carrier family. J. Mol. Biol. 1998, 277, 285–308. [Google Scholar] [CrossRef] [PubMed]
- di Punzio, G.; Di Noia, M.A.; Delahodde, A.; Sellem, C.; Donnini, C.; Palmieri, L.; Lodi, T.; Dallabona, C. A Yeast-Based Screening Unravels Potential Therapeutic Molecules for Mitochondrial Diseases Associated with Dominant ANT1 Mutations. Int. J. Mol. Sci. 2021, 22, 4461. [Google Scholar] [CrossRef]
- Aprille, J.R. Mechanism and regulation of the mitochondrial ATP-Mg/P(i) carrier. J. Bioenerg. Biomembr. 1993, 25, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J. Sal1p, a calcium-dependent carrier protein that suppresses an essential cellular function associated With the Aac2 isoform of ADP/ATP translocase in Saccharomyces cerevisiae. Genetics 2004, 167, 607–617. [Google Scholar] [CrossRef]
- Traba, J.; Froschauer, E.M.; Wiesenberger, G.; Satrustegui, J.; Del Arco, A. Yeast mitochondria import ATP through the calcium-dependent ATP-Mg/Pi carrier Sal1p, and are ATP consumers during aerobic growth in glucose. Mol. Microbiol. 2008, 69, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Cavero, S.; Traba, J.; Del Arco, A.; Satrustegui, J. The calcium-dependent ATP-Mg/Pi mitochondrial carrier is a target of glucose-induced calcium signalling in Saccharomyces cerevisiae. Biochem. J. 2005, 392, 537–544. [Google Scholar] [CrossRef]
- Laco, J.; Zeman, I.; Pevala, V.; Polčic, P.; Kolarov, J. Adenine nucleotide transport via Sal1 carrier compensates for the essential function of the mitochondrial ADP/ATP carrier. FEMS Yeast Res. 2010, 10, 290–296. [Google Scholar] [CrossRef][Green Version]
- Fiermonte, G.; De Leonardis, F.; Todisco, S.; Palmieri, L.; Lasorsa, F.M.; Palmieri, F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 2004, 279, 30722–30730. [Google Scholar] [CrossRef]
- Traba, J.; Satrustegui, J.; del Arco, A. Characterization of SCaMC-3-like/slc25a41, a novel calcium-independent mitochondrial ATP-Mg/Pi carrier. Biochem. J. 2009, 418, 125–133. [Google Scholar] [CrossRef][Green Version]
- Ryu, J.; Ko, J.M.; Shin, C.H. A 9-year-old Korean girl with Fontaine progeroid syndrome: A case report with further phenotypical delineation and description of clinical course during long-term follow-up. BMC Med. Genet. 2019, 20, 188. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, M.E.; Cotrina-Vinagre, F.J.; Cruz-Rojo, J.; Garzon-Lorenzo, L.; Carnicero-Rodriguez, P.; Pozo, J.S.; Martinez-Azorin, F. A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24. Am. J. Med. Genet. A 2018, 176, 2479–2486. [Google Scholar] [CrossRef]
- Writzl, K.; Maver, A.; Kovacic, L.; Martinez-Valero, P.; Contreras, L.; Satrustegui, J.; Castori, M.; Faivre, L.; Lapunzina, P.; van Kuilenburg, A.B.P.; et al. De Novo Mutations in SLC25A24 Cause a Disorder Characterized by Early Aging, Bone Dysplasia, Characteristic Face, and Early Demise. Am. J. Hum. Genet. 2017, 101, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Ehmke, N.; Graul-Neumann, L.; Smorag, L.; Koenig, R.; Segebrecht, L.; Magoulas, P.; Scaglia, F.; Kilic, E.; Hennig, A.F.; Adolphs, N.; et al. De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction. Am. J. Hum. Genet. 2017, 101, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Traba, J.; Del Arco, A.; Duchen, M.R.; Szabadkai, G.; Satrustegui, J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012, 19, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kucejova, B.; Li, L.; Wang, X.; Giannattasio, S.; Chen, X.J. Pleiotropic effects of the yeast Sal1 and Aac2 carriers on mitochondrial function via an activity distinct from adenine nucleotide transport. Mol. Genet. Genom. 2008, 280, 25–39. [Google Scholar] [CrossRef][Green Version]
- Harborne, S.P.; King, M.S.; Crichton, P.G.; Kunji, E.R. Calcium regulation of the human mitochondrial ATP-Mg/Pi carrier SLC25A24 uses a locking pin mechanism. Sci. Rep. 2017, 7, 45383. [Google Scholar] [CrossRef]
- Jabalameli, M.R.; Fitzpatrick, F.M.; Colombo, R.; Howles, S.A.; Leggatt, G.; Walker, V.; Wiberg, A.; Kunji, E.R.S.; Ennis, S. Exome sequencing identifies a disease variant of the mitochondrial ATP-Mg/Pi carrier SLC25A25 in two families with kidney stones. Mol. Genet. Genom. Med. 2021, e1749. [Google Scholar] [CrossRef]
- Phelps, A.; Schobert, C.T.; Wohlrab, H. Cloning and characterization of the mitochondrial phosphate transport protein gene from the yeast Saccharomyces cerevisiae. Biochemistry 1991, 30, 248–252. [Google Scholar] [CrossRef]
- Hamel, P.; Saint-Georges, Y.; de Pinto, B.; Lachacinski, N.; Altamura, N.; Dujardin, G. Redundancy in the function of mitochondrial phosphate transport in Saccharomyces cerevisiae and Arabidopsis thaliana. Mol. Microbiol. 2004, 51, 307–317. [Google Scholar] [CrossRef]
- Bhagi-Damodaran, A.; Michael, M.A.; Zhu, Q.; Reed, J.; Sandoval, B.A.; Mirts, E.N.; Chakraborty, S.; Moenne-Loccoz, P.; Zhang, Y.; Lu, Y. Why copper is preferred over iron for oxygen activation and reduction in haem-copper oxidases. Nat. Chem. 2017, 9, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Vest, K.E.; Leary, S.C.; Winge, D.R.; Cobine, P.A. Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 2013, 288, 23884–23892. [Google Scholar] [CrossRef] [PubMed]
- Dolce, V.; Fiermonte, G.; Palmieri, F. Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 1996, 399, 95–98. [Google Scholar] [CrossRef]
- Dolce, V.; Iacobazzi, V.; Palmieri, F.; Walker, J.E. The sequences of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J. Biol. Chem. 1994, 269, 10451–10460. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; Palmieri, F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 1998, 273, 22782–22787. [Google Scholar] [CrossRef] [PubMed]
- Zara, V.; Palmieri, F.; Mahlke, K.; Pfanner, N. The cleavable presequence is not essential for import and assembly of the phosphate carrier of mammalian mitochondria but enhances the specificity and efficiency of import. J. Biol. Chem. 1992, 267, 12077–12081. [Google Scholar] [CrossRef]
- Yamagoshi, R.; Yamamoto, T.; Hashimoto, M.; Sugahara, R.; Shiotsuki, T.; Miyoshi, H.; Terada, H.; Shinohara, Y. Identification of amino acid residues of mammalian mitochondrial phosphate carrier important for its functional expression in yeast cells, as achieved by PCR-mediated random mutation and gap-repair cloning. Mitochondrion 2017, 32, 1–9. [Google Scholar] [CrossRef]
- Mayr, J.A.; Zimmermann, F.A.; Horvath, R.; Schneider, H.C.; Schoser, B.; Holinski-Feder, E.; Czermin, B.; Freisinger, P.; Sperl, W. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 2011, 21, 803–808. [Google Scholar] [CrossRef]
- Mayr, J.A.; Merkel, O.; Kohlwein, S.D.; Gebhardt, B.R.; Bohles, H.; Fotschl, U.; Koch, J.; Jaksch, M.; Lochmuller, H.; Horvath, R.; et al. Mitochondrial phosphate-carrier deficiency: A novel disorder of oxidative phosphorylation. Am. J. Hum. Genet. 2007, 80, 478–484. [Google Scholar] [CrossRef]
- Halestrap, A.P. The mitochondrial pyruvate carrier: Has it been unearthed at last? Cell Metab. 2012, 16, 141–143. [Google Scholar] [CrossRef]
- Hildyard, J.C.; Halestrap, A.P. Identification of the mitochondrial pyruvate carrier in Saccharomyces cerevisiae. Biochem. J. 2003, 374, 607–611. [Google Scholar] [CrossRef]
- Todisco, S.; Agrimi, G.; Castegna, A.; Palmieri, F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.L.; Zamboni, N.; Westermann, B.; Kunji, E.R.; Martinou, J.C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012, 337, 93–96. [Google Scholar] [CrossRef]
- Tavoulari, S.; Thangaratnarajah, C.; Mavridou, V.; Harbour, M.E.; Martinou, J.C.; Kunji, E.R. The yeast mitochondrial pyruvate carrier is a hetero-dimer in its functional state. EMBO J. 2019, 38, e100785. [Google Scholar] [CrossRef] [PubMed]
- Bender, T.; Pena, G.; Martinou, J.C. Regulation of mitochondrial pyruvate uptake by alternative pyruvate carrier complexes. EMBO J. 2015, 34, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Brivet, M.; Garcia-Cazorla, A.; Lyonnet, S.; Dumez, Y.; Nassogne, M.C.; Slama, A.; Boutron, A.; Touati, G.; Legrand, A.; Saudubray, J.M. Impaired mitochondrial pyruvate importation in a patient and a fetus at risk. Mol. Genet. Metab. 2003, 78, 186–192. [Google Scholar] [CrossRef]
- Oonthonpan, L.; Rauckhorst, A.J.; Gray, L.R.; Boutron, A.C.; Taylor, E.B. Two human patient mitochondrial pyruvate carrier mutations reveal distinct molecular mechanisms of dysfunction. JCI Insight 2019, 5, e126132. [Google Scholar] [CrossRef]
- Cavero, S.; Vozza, A.; del Arco, A.; Palmieri, L.; Villa, A.; Blanco, E.; Runswick, M.J.; Walker, J.E.; Cerdan, S.; Palmieri, F.; et al. Identification and metabolic role of the mitochondrial aspartate-glutamate transporter in Saccharomyces cerevisiae. Mol. Microbiol. 2003, 50, 1257–1269. [Google Scholar] [CrossRef]
- Palmieri, L.; Agrimi, G.; Runswick, M.J.; Fearnley, I.M.; Palmieri, F.; Walker, J.E. Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J. Biol. Chem. 2001, 276, 1916–1922. [Google Scholar] [CrossRef]
- Schikora-Tamarit, M.A.; Marcet-Houben, M.; Nosek, J.; Gabaldon, T. Shared evolutionary footprints suggest mitochondrial oxidative damage underlies multiple complex I losses in fungi. Open Biol. 2021, 11, 200362. [Google Scholar] [CrossRef]
- Palmieri, F.; Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim. Biophys. Acta 2016, 1863, 2362–2378. [Google Scholar] [CrossRef]
- Kakhniashvili, D.; Mayor, J.A.; Gremse, D.A.; Xu, Y.; Kaplan, R.S. Identification of a novel gene encoding the yeast mitochondrial dicarboxylate transport protein via overexpression, purification, and characterization of its protein product. J. Biol. Chem. 1997, 272, 4516–4521. [Google Scholar] [CrossRef]
- Palmieri, L.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification by bacterial expression and functional reconstitution of the yeast genomic sequence encoding the mitochondrial dicarboxylate carrier protein. FEBS Lett. 1996, 399, 299–302. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrustegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Thangaratnarajah, C.; Ruprecht, J.J.; Kunji, E.R. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat. Commun. 2014, 5, 5491. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; Indiveri, C.; Palmieri, F. Purification of reconstitutively active alpha-oxoglutarate carrier from pig heart mitochondria. Biochim. Biophys. Acta 1985, 810, 362–369. [Google Scholar] [CrossRef]
- Runswick, M.J.; Walker, J.E.; Bisaccia, F.; Iacobazzi, V.; Palmieri, F. Sequence of the bovine 2-oxoglutarate/malate carrier protein: Structural relationship to other mitochondrial transport proteins. Biochemistry 1990, 29, 11033–11040. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Walker, J.E.; Palmieri, F. Abundant bacterial expression and reconstitution of an intrinsic membrane-transport protein from bovine mitochondria. Biochem. J. 1993, 294 Pt 1, 293–299. [Google Scholar] [CrossRef]
- Miniero, D.V.; Cappello, A.R.; Curcio, R.; Ludovico, A.; Daddabbo, L.; Stipani, I.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Functional and structural role of amino acid residues in the matrix alpha-helices, termini and cytosolic loops of the bovine mitochondrial oxoglutarate carrier. Biochim. Biophys. Acta 2011, 1807, 302–310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- del Arco, A.; Agudo, M.; Satrustegui, J. Characterization of a second member of the subfamily of calcium-binding mitochondrial carriers expressed in human non-excitable tissues. Biochem. J. 2000, 345 Pt 3, 725–732. [Google Scholar] [CrossRef] [PubMed]
- del Arco, A.; Satrustegui, J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain. J. Biol. Chem. 1998, 273, 23327–23334. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Kobayashi, K.; Iijima, M.; Horiuchi, M.; Begum, L.; Jalil, M.A.; Li, M.X.; Lu, Y.B.; Ushikai, M.; Tabata, A.; et al. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: Involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle. Mol. Genet. Metab. 2004, 81 (Suppl. 1), S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Wibom, R.; Lasorsa, F.M.; Tohonen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. JIMD Rep. 2014, 14, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Napolioni, V.; Persico, A.M.; Porcelli, V.; Palmieri, L. The mitochondrial aspartate/glutamate carrier AGC1 and calcium homeostasis: Physiological links and abnormalities in autism. Mol. Neurobiol. 2011, 44, 83–92. [Google Scholar] [CrossRef]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; von Dobeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef]
- Perez-Liebana, I.; Casarejos, M.J.; Alcaide, A.; Herrada-Soler, E.; Llorente-Folch, I.; Contreras, L.; Satrustegui, J.; Pardo, B. βOHB Protective Pathways in Aralar-KO Neurons and Brain: An Alternative to Ketogenic Diet. J. Neurosci. 2020, 40, 9293–9305. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Punzi, G.; Obre, E.; Lacombe, D.; De Grassi, A.; Pierri, C.L.; Rossignol, R. AGC1/2, the mitochondrial aspartate-glutamate carriers. Biochim. Biophys. Acta 2016, 1863, 2394–2412. [Google Scholar] [CrossRef]
- Saheki, T.; Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J. Hum. Genet. 2002, 47, 333–341. [Google Scholar] [CrossRef]
- Saheki, T.; Moriyama, M.; Funahashi, A.; Kuroda, E. AGC2 (Citrin) Deficiency-From Recognition of the Disease till Construction of Therapeutic Procedures. Biomolecules 2020, 10, 1100. [Google Scholar] [CrossRef]
- Wongkittichote, P.; Tungpradabkul, S.; Wattanasirichaigoon, D.; Jensen, L.T. Prediction of the functional effect of novel SLC25A13 variants using a S. cerevisiae model of AGC2 deficiency. J. Inherit. Metab. Dis. 2013, 36, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Lin, W.X.; Deng, M.; Zhao, S.T.; Zeng, H.S.; Chen, F.P.; Song, Y.Z. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). PLoS ONE 2014, 9, e89267. [Google Scholar] [CrossRef]
- Fiermonte, G.; Dolce, V.; Palmieri, L.; Ventura, M.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001, 276, 8225–8230. [Google Scholar] [CrossRef]
- Boczonadi, V.; King, M.S.; Smith, A.C.; Olahova, M.; Bansagi, B.; Roos, A.; Eyassu, F.; Borchers, C.; Ramesh, V.; Lochmuller, H.; et al. Mitochondrial oxodicarboxylate carrier deficiency is associated with mitochondrial DNA depletion and spinal muscular atrophy-like disease. Genet. Med. 2018, 20, 1224–1235. [Google Scholar] [CrossRef]
- Castegna, A.; Scarcia, P.; Agrimi, G.; Palmieri, L.; Rottensteiner, H.; Spera, I.; Germinario, L.; Palmieri, F. Identification and functional characterization of a novel mitochondrial carrier for citrate and oxoglutarate in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 17359–17370. [Google Scholar] [CrossRef]
- Scarcia, P.; Palmieri, L.; Agrimi, G.; Palmieri, F.; Rottensteiner, H. Three mitochondrial transporters of Saccharomyces cerevisiae are essential for ammonium fixation and lysine biosynthesis in synthetic minimal medium. Mol. Genet. Metab. 2017, 122, 54–60. [Google Scholar] [CrossRef]
- Fiermonte, G.; Palmieri, L.; Dolce, V.; Lasorsa, F.M.; Palmieri, F.; Runswick, M.J.; Walker, J.E. The sequence, bacterial expression, and functional reconstitution of the rat mitochondrial dicarboxylate transporter cloned via distant homologs in yeast and Caenorhabditis elegans. J. Biol. Chem. 1998, 273, 24754–24759. [Google Scholar] [CrossRef] [PubMed]
- Punzi, G.; Porcelli, V.; Ruggiu, M.; Hossain, M.F.; Menga, A.; Scarcia, P.; Castegna, A.; Gorgoglione, R.; Pierri, C.L.; Laera, L.; et al. SLC25A10 biallelic mutations in intractable epileptic encephalopathy with complex I deficiency. Hum. Mol. Genet. 2018, 27, 499–504. [Google Scholar] [CrossRef]
- Palmieri, L.; Vozza, A.; Honlinger, A.; Dietmeier, K.; Palmisano, A.; Zara, V.; Palmieri, F. The mitochondrial dicarboxylate carrier is essential for the growth of Saccharomyces cerevisiae on ethanol or acetate as the sole carbon source. Mol. Microbiol. 1999, 31, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.S.; Mayor, J.A.; Gremse, D.A.; Wood, D.O. High level expression and characterization of the mitochondrial citrate transport protein from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 4108–4114. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Drexler, K.; Federlin, M.; Schlitt, H.J.; Berneburg, M.; Adamski, J.; Gaumann, A.; Geissler, E.K.; Ganapathy, V.; Parkinson, E.K.; et al. Extracellular Citrate Fuels Cancer Cell Metabolism and Growth. Front. Cell Dev. Biol. 2020, 8, 602476. [Google Scholar] [CrossRef]
- Nota, B.; Struys, E.A.; Pop, A.; Jansen, E.E.; Fernandez Ojeda, M.R.; Kanhai, W.A.; Kranendijk, M.; van Dooren, S.J.; Bevova, M.R.; Sistermans, E.A.; et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am. J. Hum. Genet. 2013, 92, 627–631. [Google Scholar] [CrossRef]
- Pop, A.; Williams, M.; Struys, E.A.; Monne, M.; Jansen, E.E.W.; De Grassi, A.; Kanhai, W.A.; Scarcia, P.; Ojeda, M.R.F.; Porcelli, V.; et al. An overview of combined D-2- and L-2-hydroxyglutaric aciduria: Functional analysis of CIC variants. J. Inherit. Metab. Dis. 2018, 41, 169–180. [Google Scholar] [CrossRef]
- Edvardson, S.; Porcelli, V.; Jalas, C.; Soiferman, D.; Kellner, Y.; Shaag, A.; Korman, S.H.; Pierri, C.L.; Scarcia, P.; Fraenkel, N.D.; et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutations in SLC25A1 encoding the mitochondrial citrate transporter. J. Med. Genet. 2013, 50, 240–245. [Google Scholar] [CrossRef]
- Girardi, E.; Agrimi, G.; Goldmann, U.; Fiume, G.; Lindinger, S.; Sedlyarov, V.; Srndic, I.; Gurtl, B.; Agerer, B.; Kartnig, F.; et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat. Commun. 2020, 11, 6145. [Google Scholar] [CrossRef]
- Kory, N.; Uit de Bos, J.; van der Rijt, S.; Jankovic, N.; Gura, M.; Arp, N.; Pena, I.A.; Prakash, G.; Chan, S.H.; Kunchok, T.; et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci. Adv. 2020, 6, eabe5310. [Google Scholar] [CrossRef]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef]
- Nosaka, K.; Kaneko, Y.; Nishimura, H.; Iwashima, A. Isolation and characterization of a thiamin pyrophosphokinase gene, THI80, from Saccharomyces cerevisiae. J. Biol. Chem. 1993, 268, 17440–17447. [Google Scholar] [CrossRef]
- Hohmann, S.; Meacock, P.A. Thiamin metabolism and thiamin diphosphate-dependent enzymes in the yeast Saccharomyces cerevisiae: Genetic regulation. Biochim. Biophys. Acta 1998, 1385, 201–219. [Google Scholar] [CrossRef]
- Marobbio, C.M.; Vozza, A.; Harding, M.; Bisaccia, F.; Palmieri, F.; Walker, J.E. Identification and reconstitution of the yeast mitochondrial transporter for thiamine pyrophosphate. EMBO J. 2002, 21, 5653–5661. [Google Scholar] [CrossRef]
- Chipman, D.; Barak, Z.; Schloss, J.V. Biosynthesis of 2-aceto-2-hydroxy acids: Acetolactate synthases and acetohydroxyacid synthases. Biochim. Biophys. Acta 1998, 1385, 401–419. [Google Scholar] [CrossRef]
- Dolce, V.; Fiermonte, G.; Runswick, M.J.; Palmieri, F.; Walker, J.E. The human mitochondrial deoxynucleotide carrier and its role in the toxicity of nucleoside antivirals. Proc. Natl. Acad. Sci. USA 2001, 98, 2284–2288. [Google Scholar] [CrossRef]
- Lindhurst, M.J.; Fiermonte, G.; Song, S.; Struys, E.; De Leonardis, F.; Schwartzberg, P.L.; Chen, A.; Castegna, A.; Verhoeven, N.; Mathews, C.K.; et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc. Natl. Acad. Sci. USA 2006, 103, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.I.; Robinson, D.; Puffenberger, E.G.; Strauss, K.A.; Morton, D.H. Amish lethal microcephaly: A new metabolic disorder with severe congenital microcephaly and 2-ketoglutaric aciduria. Am. J. Hum. Genet. 2002, 112, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.J.; Agarwala, R.; Bouffard, G.; Davis, J.; Fiermonte, G.; Hilliard, M.S.; Koch, T.; Kalikin, L.M.; Makalowska, I.; Morton, D.H.; et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat. Genet. 2002, 32, 175–179. [Google Scholar] [CrossRef]
- Spiegel, R.; Shaag, A.; Edvardson, S.; Mandel, H.; Stepensky, P.; Shalev, S.A.; Horovitz, Y.; Pines, O.; Elpeleg, O. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann. Neurol. 2009, 66, 419–424. [Google Scholar] [CrossRef]
- Goutieres, F.; Aicardi, J. Acute neurological dysfunction associated with destructive lesions of the basal ganglia in children. Ann. Neurol. 1982, 12, 328–332. [Google Scholar] [CrossRef]
- Zevit, N.; Steinmetz, A.; Kornreich, L.; Straussberg, R. Acute infantile bilateral striatal necrosis: Single-photon emission computed tomography (SPECT) imaging and review. J. Child. Neurol. 2007, 22, 1222–1226. [Google Scholar] [CrossRef]
- Palmieri, F.; Scarcia, P.; Monne, M. Diseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review. Biomolecules 2020, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Agarwal, S.; Heyman, J.A.; Matson, S.; Heidtman, M.; Piccirillo, S.; Umansky, L.; Drawid, A.; Jansen, R.; Liu, Y.; et al. Subcellular localization of the yeast proteome. Genes Dev. 2002, 16, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.; Agrimi, G.; Lasorsa, F.M.; Palmieri, F. Identification and functional reconstitution of yeast mitochondrial carrier for S-adenosylmethionine. EMBO J. 2003, 22, 5975–5982. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sanyal, I.; Bulboaca, G.H.; Rich, A.; Flint, D.H. The gene for biotin synthase from Saccharomyces cerevisiae: Cloning, sequencing, and complementation of Escherichia coli strains lacking biotin synthase. Arch. Biochem. Biophys. 1994, 309, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sulo, P.; Martin, N.C. Isolation and characterization of LIP5. A lipoate biosynthetic locus of Saccharomyces cerevisiae. J. Biol. Chem. 1993, 268, 17634–17639. [Google Scholar] [CrossRef]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef]
- Kishita, Y.; Pajak, A.; Bolar, N.A.; Marobbio, C.M.; Maffezzini, C.; Miniero, D.V.; Monne, M.; Kohda, M.; Stranneheim, H.; Murayama, K.; et al. Intra-mitochondrial Methylation Deficiency Due to Mutations in SLC25A26. Am. J. Hum. Genet. 2015, 97, 761–768. [Google Scholar] [CrossRef]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Palmieri, L.; Porcelli, V.; Palmieri, F. Mitochondrial transporters for ornithine and related amino acids: A review. Amino Acids 2015, 47, 1763–1777. [Google Scholar] [CrossRef]
- Palmieri, L.; De Marco, V.; Iacobazzi, V.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification of the yeast ARG-11 gene as a mitochondrial ornithine carrier involved in arginine biosynthesis. FEBS Lett. 1997, 410, 447–451. [Google Scholar] [CrossRef]
- Davis, R.H. Compartmental and regulatory mechanisms in the arginine pathways of Neurospora crassa and Saccharomyces cerevisiae. Microbiol. Rev. 1986, 50, 280–313. [Google Scholar] [CrossRef]
- Crabeel, M.; Soetens, O.; De Rijcke, M.; Pratiwi, R.; Pankiewicz, R. The ARG11 gene of Saccharomyces cerevisiae encodes a mitochondrial integral membrane protein required for arginine biosynthesis. J. Biol. Chem. 1996, 271, 25011–25018. [Google Scholar] [CrossRef]
- Delforge, J.; Messenguy, F.; Wiame, J.M. The regulation of arginine biosynthesis in Saccharomyces cerevisiae. The specificity of argR- mutations and the general control of amino-acid biosynthesis. Eur. J. Biochem. 1975, 57, 231–239. [Google Scholar] [CrossRef]
- Camacho, J.A.; Obie, C.; Biery, B.; Goodman, B.K.; Hu, C.A.; Almashanu, S.; Steel, G.; Casey, R.; Lambert, M.; Mitchell, G.A.; et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat. Genet. 1999, 22, 151–158. [Google Scholar] [CrossRef]
- Morizono, H.; Woolston, J.E.; Colombini, M.; Tuchman, M. The use of yeast mitochondria to study the properties of wild-type and mutant human mitochondrial ornithine transporter. Mol. Genet. Metab. 2005, 86, 431–440. [Google Scholar] [CrossRef]
- Ersoy Tunali, N.; Marobbio, C.M.; Tiryakioglu, N.O.; Punzi, G.; Saygili, S.K.; Onal, H.; Palmieri, F. A novel mutation in the SLC25A15 gene in a Turkish patient with HHH syndrome: Functional analysis of the mutant protein. Mol. Genet. Metab. 2014, 112, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Pierri, C.L. Structure and function of mitochondrial carriers-role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef]
- Doimo, M.; Lopreiato, R.; Basso, V.; Bortolotto, R.; Tessa, A.; Santorelli, F.M.; Trevisson, E.; Salviati, L. Heterologous Expression in Yeast of Human Ornithine Carriers ORNT1 and ORNT2 and of ORNT1 Alleles Implicated in HHH Syndrome in Humans. JIMD Rep. 2016, 28, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J.A.; Rioseco-Camacho, N.; Andrade, D.; Porter, J.; Kong, J. Cloning and characterization of human ORNT2: A second mitochondrial ornithine transporter that can rescue a defective ORNT1 in patients with the hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, a urea cycle disorder. Mol. Genet. Metab. 2003, 79, 257–271. [Google Scholar] [CrossRef]
- Monné, M.; Miniero, D.V.; Daddabbo, L.; Robinson, A.J.; Kunji, E.R.; Palmieri, F. Substrate specificity of the two mitochondrial ornithine carriers can be swapped by single mutation in substrate binding site. J. Biol. Chem. 2012, 287, 7925–7934. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.; Punzi, G.; Pierri, C.L.; Palmieri, L.; Calvello, R.; Panaro, M.A.; Palmieri, F. Pathogenic potential of SLC25A15 mutations assessed by transport assays and complementation of Saccharomyces cerevisiae ORT1 null mutant. Mol. Genet. Metab. 2015, 115, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, V.; Fiermonte, G.; Longo, A.; Palmieri, F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J. Biol. Chem. 2014, 289, 13374–13384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Q.; Gu, J.; Yin, L.; Liang, S.; Wu, L.; Xu, H.; Zhao, C.; Gu, Y. Elevated mitochondrial SLC25A29 in cancer modulates metabolic status by increasing mitochondria-derived nitric oxide. Oncogene 2018, 37, 2545–2558. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J.A.; Rioseco-Camacho, N. The human and mouse SLC25A29 mitochondrial transporters rescue the deficient ornithine metabolism in fibroblasts of patients with the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr. Res. 2009, 66, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kunau, W.H.; Dommes, V.; Schulz, H. beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: A century of continued progress. Prog. Lipid. Res. 1995, 34, 267–342. [Google Scholar] [CrossRef]
- van Roermund, C.W.; Hettema, E.H.; van den Berg, M.; Tabak, H.F.; Wanders, R.J. Molecular characterization of carnitine-dependent transport of acetyl-CoA from peroxisomes to mitochondria in Saccharomyces cerevisiae and identification of a plasma membrane carnitine transporter, Agp2p. EMBO J. 1999, 18, 5843–5852. [Google Scholar] [CrossRef]
- Palmieri, L.; Lasorsa, F.M.; Iacobazzi, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae. FEBS Lett. 1999, 462, 472–476. [Google Scholar] [CrossRef]
- Huizing, M.; Iacobazzi, V.; Ijlst, L.; Savelkoul, P.; Ruitenbeek, W.; van den Heuvel, L.; Indiveri, C.; Smeitink, J.; Trijbels, F.; Wanders, R.; et al. Cloning of the human carnitine-acylcarnitine carrier cDNA and identification of the molecular defect in a patient. Am. J. Hum. Genet. 1997, 61, 1239–1245. [Google Scholar] [CrossRef]
- IJlst, L.; van Roermund, C.W.; Iacobazzi, V.; Oostheim, W.; Ruiter, J.P.; Williams, J.C.; Palmieri, F.; Wanders, R.J. Functional analysis of mutant human carnitine acylcarnitine translocases in yeast. Biochem. Biophys. Res. Commun. 2001, 280, 700–706. [Google Scholar] [CrossRef]
- Prohl, C.; Pelzer, W.; Diekert, K.; Kmita, H.; Bedekovics, T.; Kispal, G.; Lill, R. The yeast mitochondrial carrier Leu5p and its human homologue Graves’ disease protein are required for accumulation of coenzyme A in the matrix. Mol. Cell Biol. 2001, 21, 1089–1097. [Google Scholar] [CrossRef]
- Palmieri, L.; Lasorsa, F.M.; De Palma, A.; Palmieri, F.; Runswick, M.J.; Walker, J.E. Identification of the yeast ACR1 gene product as a succinate-fumarate transporter essential for growth on ethanol or acetate. FEBS Lett. 1997, 417, 114–118. [Google Scholar] [CrossRef]
- Palmieri, L.; Vozza, A.; Agrimi, G.; De Marco, V.; Runswick, M.J.; Palmieri, F.; Walker, J.E. Identification of the yeast mitochondrial transporter for oxaloacetate and sulfate. J. Biol. Chem. 1999, 274, 22184–22190. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Damiano, F.; De Benedetto, G.; Siculella, L.; Pennetta, A.; Muto, L.; Paradies, E.; Marobbio, C.M.; Dolce, V.; Capobianco, L. Characterization of Human and Yeast Mitochondrial Glycine Carriers with Implications for Heme Biosynthesis and Anemia. J. Biol. Chem. 2016, 291, 19746–19759. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, A.; Jang, J.; Glerum, D.M.; Wu, M. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J. Biol. Chem. 1996, 271, 7392–7397. [Google Scholar] [CrossRef]
- Vozza, A.; Blanco, E.; Palmieri, L.; Palmieri, F. Identification of the mitochondrial GTP/GDP transporter in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 20850–20857. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.; Di Noia, M.A.; Palmieri, F. Identification of a mitochondrial transporter for pyrimidine nucleotides in Saccharomyces cerevisiae: Bacterial expression, reconstitution and functional characterization. Biochem. J. 2006, 393, 441–446. [Google Scholar] [CrossRef][Green Version]
- Todisco, S.; Di Noia, M.A.; Castegna, A.; Lasorsa, F.M.; Paradies, E.; Palmieri, F. The Saccharomyces cerevisiae gene YPR011c encodes a mitochondrial transporter of adenosine 5′-phosphosulfate and 3′-phospho-adenosine 5′-phosphosulfate. Biochim. Biophys. Acta 2014, 1837, 326–334. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Penmatsa, A.; Whittaker, J.W. The Mtm1p carrier and pyridoxal 5′-phosphate cofactor trafficking in yeast mitochondria. Arch. Biochem. Biophys. 2015, 568, 64–70. [Google Scholar] [CrossRef]
- Froschauer, E.M.; Schweyen, R.J.; Wiesenberger, G. The yeast mitochondrial carrier proteins Mrs3p/Mrs4p mediate iron transport across the inner mitochondrial membrane. Biochim. Biophys. Acta 2009, 1788, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, S.; Wang, J.; Wang, X.; Gao, B.; Fan, Q.; Sun, F.; Zhou, B. A novel mitochondrial carrier protein Mme1 acts as a yeast mitochondrial magnesium exporter. Biochim. Biophys. Acta 2015, 1853, 724–732. [Google Scholar] [CrossRef][Green Version]
- Sesaki, H.; Jensen, R.E. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J. Cell. Biol. 2001, 152, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, L.; Rottensteiner, H.; Girzalsky, W.; Scarcia, P.; Palmieri, F.; Erdmann, R. Identification and functional reconstitution of the yeast peroxisomal adenine nucleotide transporter. EMBO J. 2001, 20, 5049–5059. [Google Scholar] [CrossRef] [PubMed]
- van Roermund, C.W.; Drissen, R.; van Den Berg, M.; IJlst, L.; Hettema, E.H.; Tabak, H.F.; Waterham, H.R.; Wanders, R.J. Identification of a peroxisomal ATP carrier required for medium-chain fatty acid beta-oxidation and normal peroxisome proliferation in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 4321–4329. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Mitochondrial Carrier | Yeast Genes 1 | Human Genes 1 | Human Disease (OMIM) 2 |
|---|---|---|---|
| ADP/ATP | AAC1 AAC2 (PET9) AAC3 | ANT1 (SLC25A4) ANT2 (SLC25A5) ANT3 (SLC25A6) ANT4 (SLC25A31) | autosomal dominant progressive external ophthalmoplegia with mtDNA deletions (adPEO) (609283); mtDNA depletion syndrome 12a (cardiomyopathic type), autosomal dominant, MTDPS12a (617184); mtDNA depletion syndrome 12b (cardiomyopathic type), autosomal recessive, MTDPS12b (615418) |
| ATP-Mg/Pi | SAL1 | APC1 (SCaMC-1, SLC25A24) APC2 (SCaMC3, SLC25A23) APC3 (SCaMC2, SLC25A25) SCaMC-3-like (SLC25A41) | Fontaine progeroid syndrome (612289) |
| phosphate | MIR1 PIC2 | PiC (SLC25A3) | mitochondrial phosphate carrier deficiency, MPCD (610773) |
| thiamine pyrophosphate | TPC1 | TPC (SLC25A19) | Amish microcephaly (607196); bilateral striatal necrosis (613710) |
| ornithine/citrulline | ORT1 (ARG11) | ORC1 (Ornt1, SLC25A15) ORC2 (Ornt2, SLC25A2) | hyperornithinaemia-hyperammonaemia-homocitrullinuria (238970) |
| aspartate/glutamate | AGC1 | AGC1 (aralar1, SLC25A12) AGC2 (citrin, SLC25A13) | developmental and epileptic encephalopathy 39 (612949); neonatal onset type 2 citrullinaemia (NiCCD) (605814); adult-onset type 2 citrullinaemia (CTLN2) (603471) |
| oxodicarboxylate | ODC1 ODC2 | ODC (SLC25A21) | mtDNA depletion syndrome and spinal muscular atrophy-like disease (618811) |
| dicarboxylate | DIC1 | DIC (SLC25A10) | intractable epileptic encephalopathy with complex I deficiency |
| citrate | CTP1 | CIC (SLC25A1) | combined d-2- and l-2-hydroxyglutaric aciduria (615182) |
| NAD+ | NDT1 NDT2 (YEA6) | SLC25A51 (MCART1) SLC25A52 (MCART2) | |
| S-adenosylmethionine | SAM5 (PET8) | SAMC (SLC25A26) | combined oxidative phosphorylation deficiency 28, COXPD28 (616794) |
| coenzyme A | LEU5 | SLC25A42 | recurrent metabolic crises with variable encephalomyopathic features and neurologic regression, MECREN (618416) |
| succinate/fumarate | SFC1 (ACR1) | ||
| citrate/oxoglutarate | YHM2 | ||
| oxaloacetate | OAC1 | ||
| carnitine | CRC1 | CAC (SLC25A20) | carnitine-acylcarnitine translocase deficiency (212138) |
| glycine | HEM25 YMC1? | GlyC (SLC25A38) | sideroblastic anemia-2 (SIDBA2), (pyridoxine-refractory) (205950) |
| glutamate | YMC2 | GC1 (SLC25A22) GC2 (SLC25A18) | developmental and epileptic encephalopathy 3 (609304) |
| FAD | FLX1 | ||
| GTP/GDP | GGC1 (YHM1, SHM1) | ||
| pyrimidine nucleotides | RIM2 | PNC1 (SLC25A33) PNC2 (SLC25A36) | |
| APS, PAPS 3 | MRX21 | SLC25A42 | |
| Fe2+ | MRS3 MRS4 | MFRN1 (SLC25A37) MFRN2 (SLC25A28) | |
| Mg2+ exporter | MME1 | ||
| pyridoxal 5′-phosphate | MTM1 | ||
| uncharacterized | MRX20 | ||
| ATP/AMP | ANT14 | ||
| UGO15 | |||
| pyruvate 6 | MPC1 MPC2 MPC3 | MPC1 MPC2 | mitochondrial pyruvate carrier deficiency, MPYCD (614741) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mentel, M.; Chovančíková, P.; Zeman, I.; Polčic, P. Learning from Yeast about Mitochondrial Carriers. Microorganisms 2021, 9, 2044. https://doi.org/10.3390/microorganisms9102044
Mentel M, Chovančíková P, Zeman I, Polčic P. Learning from Yeast about Mitochondrial Carriers. Microorganisms. 2021; 9(10):2044. https://doi.org/10.3390/microorganisms9102044
Chicago/Turabian StyleMentel, Marek, Petra Chovančíková, Igor Zeman, and Peter Polčic. 2021. "Learning from Yeast about Mitochondrial Carriers" Microorganisms 9, no. 10: 2044. https://doi.org/10.3390/microorganisms9102044
APA StyleMentel, M., Chovančíková, P., Zeman, I., & Polčic, P. (2021). Learning from Yeast about Mitochondrial Carriers. Microorganisms, 9(10), 2044. https://doi.org/10.3390/microorganisms9102044