1. Introduction
Carotenoids are terpenoid pigments with essential roles in photosynthesis in autotrophic species, but they are also produced by some heterotrophic ones [
1]. The latter include many fungi, which can produce and accumulate high levels of carotenoids [
2]. This ability is of biotechnological importance, because some carotenoids perform critical functions in animals and humans, being the source of different biological molecules and having beneficial antioxidative properties [
3,
4]. Because animals and human are not able to synthetize carotenoids, they must obtain them in the diet [
5].
The filamentous fungus
Fusarium fujikuroi, a reference model for the research of fungal carotenogenesis, produces mainly a carboxylic xanthophyll called neurosporaxanthin (NX) [
6]. The biosynthesis pathway of carotenoids in this fungus is well known, and all of the structural genes involved, called
car genes, have been previously described. NX biosynthesis in
Fusarium is induced by light and by nitrogen starvation, and both inducing effects are additive. The stimulation by light, achieved through the transcriptional induction of the structural
car genes, has been investigated in detail [
7]. In turn, the pathway is downregulated by the protein CarS, belonging to the RING finger family, and identified through the genetic characterization of
carS mutants [
8]. These mutants exhibit deep orange pigmentation under all culture conditions and accumulate large amounts of NX due to a strong upregulation of the structural
car genes regardless of light [
9,
10]. Regulation by CarS especially affects the
car cluster, consisting of the genes
carO,
carB,
carRA, and
carX. While
carB and
carRA encode two key enzymes of carotenoid biosynthesis, phytoene desaturase and phytoene synthase/carotene cyclase,
carO and
carX, encode a rhodopsin and a β-carotene cleaving enzyme, producing the CarO cofactor, retinal. The molecular mechanism of action of CarS is still not understood, but its similarity with other proteins with RING finger domains suggests a capacity to interact with E3 ligase-type enzymes that mediate ubiquitylation of target proteins. In fact, a RING finger protein, CrgA, represses carotenogenesis in
Mucor circinelloides and its lack of function causes an over-accumulation of carotenoids [
11]. Four orthologous CrgA genes were found in
Phycomyces blakesleeanus, but only one of them could complement the
crgA mutant of
M. circinelloides [
12].
The current hypothesis regarding the activity of CarS is focused on its possible interaction with other regulatory proteins to modulate their activity. Regulatory scenarios involving protein–protein interactions are not uncommon in the control of carotenogenesis in other microorganisms, even taxonomically distant, as exemplified by the complex regulatory network for the induction of carotenoid biosynthesis by light in myxobacteria [
13]. In
Myxobacterium xanthus, CarA and CarH are repressors that bind to the operator of a carotenoid operon in the dark. A third regulatory protein, CarS, acts as an antirepressor in the light, binds to CarA and CarH and disassembles them. The
F. fujikuroi CarS protein has no structural relationship with the
M. xanthus homonym, but it probably coincides in its ability to specifically interact with transcription factors involved in the control of structural
car genes. CarH is homologous of LitR, a repressor widely distributed in nonphototrophic bacteria. Proteins of the CarH/LitR family play an important role as negative regulators of light-inducible carotenoid transcription genes and at the same time serve as photosensors [
14]. Another repressor found in
Corynebacterium glutamicum and
Actinobacteria is CrtR, whose mutation causes the constitutive production of carotenoids independently of light [
15,
16]. CrtR represses its own gene and the
crt operon by binding to the promoter sequence [
15,
16]. The activity of this repressor is modulated by geranylgeranyl pyrophosphate (GGPP), which means that it is capable of sensing the amount of this metabolite [
17].
A recent RNAseq study revealed that
carS mRNA levels are low under standard laboratory conditions, especially in the dark [
18]. The objective of this work is to throw more light on CarS function through the overexpression of the
carS gene using two strategies: (i)
carS control by an inducible Tet-on expression system, and (ii)
carS overexpression by the strong P
gpdA promoter. Tet-on is an established bacterial regulatory system that was adapted to filamentous fungi to manipulate gene expression in
Aspergillus fumigatus [
19], and subsequently developed and improved in
Aspergillus niger [
20,
21]. Its mechanism is displayed in
Figure 1.
Briefly, the
A. nidulans P
gpdA promoter supports the constitutive expression of the tetracycline-dependent transactivator rtTA2
S-M2. When rtTA2
S-M2 is attached to the inducer doxycycline (Dox), it is able to bind to the operator sequence
tetO7 and activate the fungal promoter Pmin (a short version of P
gpdA) and initiate the expression of the reporter gene
mluc that encodes for the enzyme luciferase. In the presence of its substrate, luciferin, the luciferase emits light and produces oxyluciferin. This system has become a useful tool to control gene expression in many fungi [
22,
23], and was successfully used in
F. fujikuroi to activate the silent trichosetin gene cluster [
24]. Here, we used the Tet-on system to generate
F. fujikuroi strains with tunable
carS gene expression, which allowed to confirm the repressive role of this protein in carotenoid biosynthesis. The enhanced expression of
carS through Tet-on or through the constitutive P
gpdA promoter from
A. nidulans resulted in an albino phenotype, with very low carotenoid production under illumination, indicating that
carS expression is adapted to low levels for appropriate regulation of carotenoid biosynthesis in
Fusarium.
2. Materials and Methods
2.1. Strains and Culture Conditions
The wild strain of
Fusarium fujikuroi IMI58289 was obtained from the Imperial Mycological Institute (Kew, Surrey, England), and carotenoid overproducer mutant SG39 was isolated from IMI58289 by chemical mutagenesis [
8]. Both strains and transformants from this work are listed in
Table 1.
The strains were grown in DG medium, composed of 30 g glucose, 3 g NaNO
3, 1 g KH2PO4, 0.5 g KCl, 0.5 g MgSO
4·7H
2O and 2 mL of microelements [
25] per liter. Microelement are composed of 0.1 g FeSO
4·7H
2O, 0.015 g CuSO
4·5H
2O, 0.161 g ZnSO
4·7H
2O, 0.01-g MnSO
4·7H
2O, 0.01 g (NH
4)6Mo
7O
20·4H2O in 100 mL of distilled water.
Strains were cultured at 30 °C for phenotypic and molecular analysis. For sporulation, they were grown in EG agar medium in Petri dishes and incubated under white light for 7 days at 26 °C. Composition of EG medium is 1 g glucose, 1 g yeast extract, 1 g NO
3NH
4, 1 g KH
2PO
4, 0.5-g MgSO
4·7H
2O and 16 g agar per liter [
18]. Spores were harvested with water, separated from mycelia by filtration, and counted in a hemocytometer (Bürker chamber, Blau Brand, Germany). For luminescence assay, the strains were grown in DGpep, consisting of DG medium with 2 g/L of peptone.
For expression analysis, 100 mL of DG medium were inoculated with 106 fresh spores of the corresponding strain in 500-mL flaks and incubated for 3 days in an orbital shaker at 150 rpm in dark. After this time, liquid cultures were distributed in four Petri dishes, and they were exposed to white light or incubated in the dark for 1 h, with a previous adaptation to the Petri dishes for 4 h in darkness. Illumination was performed under a platform with 4 fluorescent tubes (Philips TL-D 18 W/840) at ca. 60 cm, providing a light intensity of 7 W/m2 (420 Lm/w). For DNA isolation, cultures were incubated in a similar way, without specific illumination conditions. The mycelia were collected by filtration, frozen in liquid nitrogen, and stored at −80 °C until use.
For carotenoid determinations, the strains were grown at 30 °C on 25 mL of DG agar medium in standard Petri dishes for 7 days under illumination or in darkness. Strains were inoculated with sterile toothpicks at 7 symmetrical points on each Petri dish.
2.2. DNA Isolation and PCR Assays
Genomic DNA extractions were performed using the GenElute Plant Genomic DNA Miniprep kit (Sigma-Aldrich, St. Louis, MO, USA) following the manufacturer’s instructions. DNA quality was checked by gel electrophoresis and quantified in a Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Two different DNA polymerases were used in PCR reactions. High fidelity DNA polymerase velocity (Bioline, Memphis, TN, USA) was used for plasmid construction and sequencing, while DNA polymerase BIOTAQ™ (Bioline GmbH, Germany) was used to check constructs and transformant candidates.
2.3. Generation of TETluc and TETcarS Transformants
To generate TET
luc transformants, wild protoplasts were transformed with plasmids pVG3 (P
gpdA::rtTA::T
cgrA–
tetO7::Pmin::
mluc::T
trpC) [
20] digested with
KpnI, and PAN7-1, which contains the hygromycin resistance cassette, with
hph gene, as a selection marker. After single-spore purification, we obtained three transformants which were analyzed by PCR, to check pVG3 integration, using two different combinations of internal primer pairs (
Table S1). The three transformants gave a 1-kb band with primers Pmin-1F and mluc-1R and a band of 2.3 kb with rtTA2-1F and mluc-1R primers, indicating the integration of the Tet-on cassette (
Figure S1).
To generate TET
carS transformants, protoplasts from wild-type and SG39 mycelia were transformed with the linear pPO5 and the PAN7-1 plasmids, with the hygromycin resistance cassette [
26], as described [
27]. After the purification procedure in hygromycin-supplemented medium, two different kinds of transformants were obtained, seven originating from the wild strain and nine from the
carS mutant strain. Both kinds of transformants were checked by PCR using the same pair of primers, Pmin-1F and CarS-9R, which should amplify a 1.6-kb band (
Table S1, Figures S2 and S3). Three transformants from the
carS mutant (T6, T7, T9) and four from the wild strain (T1, T5, T6, T7) showed the correct band and were considered as positive transformants.
2.4. Construction of pJM2 Plasmid and Generation of carS Constitutive Overexpression Transformants
For
carS overexpression, the
carS coding sequence was placed under the control of
gpdA promoter (P
gpdA). The P
gpdA::carS fragment was generated by fusion PCR. P
gpdA was amplified from PAN7-1 vector using primers PgpdA-Not-1F and PgpdA-carS-1R. The forward primer contained a
NotI restriction site and reverse primer PgpdA-carS-1R included an overlapping sequence of 21 pb with the start codon of
carS (
Table S1). A 2.2-kb DNA fragment, containing the
carS gene and part of the
trpC terminator, was amplified from plasmid pPO5 using primers carS-PmeI-1F and trpC-MluI-2R with restriction sites for
PmeI and
MluI, respectively. Both fragments were fused with primers PgpdA-Not-1F and trpC-MluI-2R. The fusion product was purified and ligated to pGEM
®-T easy vector (Promega), resulting in pJM1 plasmid. For pJM2 construction, plasmids pJM1 and PBN008 (containing the
amdS cassette allowing the use of acetamide as nitrogen source) were digested with
NotI and
MluI restriction enzymes, and the purified DNA fragment (P
gpdA::carS) was ligated to the digested PBN008 plasmid.
Wild protoplasts were transformed with the pJM2 plasmid following our standard protocol [
27], and three transformants were isolated after the single-spore purification procedure in the selective medium with acetamide as nitrogen source. Transformants were analyzed by PCR, using primers S1carS-1F and amiE-1R that should amplify a 1.7-kb band that is found in all the candidates (
Figure S4).
2.5. Southern Blot Analyses
Southern blot analyses were conducted using digoxigenin-labelled probes, which were amplified by PCR from genomic DNA and labelled with the DIG DNA Labelling Mix (Roche, Mannheim, Germany), following the manufacturer’s instructions. The sensitivity of the probes was checked before their use. Approximately 15 μg of genomic DNA from transformants were digested with different enzymes, electrophoresed in 0.7% agarose gels, and transferred by capillarity to a positively charged nylon membrane (Hybond-N from Amersham) as described [
28]. The membrane was incubated with 25 mL of prehybridization solution: 5x Saline Sodium Citrate (5x SSC, containing 750 mM NaCl in 75-mM sodium citrate, pH 7.0), 0.7% sodium dodecyl sulfate (SDS), 0.1% N-lauroylsarcosine, and 2x Roche blocking reagent in maleic buffer (118.3 mM maleic acid, 150 mM NaCl, pH 7.5) at 50 °C in a glass cylinder for 1 h in a hybridization oven (HB-100 Hybridizer, UVP). After this, it was incubated overnight with the same solution containing 25 ng/mL of probe.
Afterwards, the membrane was washed twice with 2x washing solution: 2x SSC and 0.1% SDS for 5 min at 25 °C, and twice with 0.1x washing solution: 0.1x SSC and 0.1% SDS at 68 °C. After equilibrating the nylon with maleic buffer, it was blocked for 1 h. Then, 3-μL of antibody antidigoxigenin (Anti-Digoxigenin-AP, Fab fragments, Roche, Mannheim, Germany) was added to 30 mL of blocking solution and incubated for 30 min. The membrane was then washed twice with maleic buffer with 0.3% Tween-20 for 15 min and incubated between acetates for 2 min at room temperature in detection buffer. Detection was performed with CDP-Star®, ready-to-use (Roche), and signals were detected in an Odyssey Fc Imaging System (LI-COR, Lincoln, NE, USA).
2.6. Luminescence Assay
Luminescence emission from TETluc transformants (SG253 and SG255) was measured in Costar 96-well white clear-bottom plates (Corning, Corning, NY, USA) using the wild strain as a negative control. In each well, 50 μL of 2x DGpep was inoculated with 104 fresh spores and incubated at 30 °C for 16 h. Afterwards, 50 μL of doxycycline, hereafter Dox, was added at different concentrations (2.5, 5, 10 and 20 μg/mL in water) to activate rtTA2S-M2.
A total of 50 μL of 0.2 mM luciferin (Promega, Madison, WI, USA), previously diluted in 2x DGpep medium, was added to each well with a final volume of 200 μL. For luminescence detection, measurements were periodically taken in a Multimodal Synergy HT plate lector for 55 h, and the absorbance at 600 nm was simultaneously measured. Data were analyzed in reference to absorbance.
2.7. Expression Analyses
All the strains were cultured in the dark except for the OEcarS transformants, which were also exposed to light. Total RNA was isolated using the RNeasy Plant Mini Kit (Qiagen, Chatsworth, CA, USA). RNA samples were checked by electrophoresis and quantified by Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Retrotranscription to cDNA was made with 2.5 μg of RNA, using the Transcriptor first-strand cDNA synthesis kit (Roche, Mannheim, Germany), and final cDNA concentrations were adjusted to 25 ng/μL.
The RT-PCR measurements were performed in a LightCycler 480 real-time instrument (Roche Mannheim, Germany) with the LightCycler 480 SYBR green I Master kit (Roche, Mannheim, Germany), following the manufacturer’s protocols. The primers used for the amplification and detection of the mRNA of genes
carS,
carB,
carRA,
mluc, FFUJ_04397 and
gpdA are listed in
Table S2. Expression values were normalized against those of the reference β
1 tubulin and
gpdA genes.
2.8. Carotenoid Measurements
For quantification of carotenoids, the strains were grown on DG agar medium, and in the case of the Tet-on transformants, the media also contained 20 µg/mL Dox, except for the control plates. After seven days of incubation at 30 °C under illumination, mycelia were removed from agar media with a clean scalpel blade and stored at 20 °C.
Carotenoids were extracted with acetone from lyophilized mycelia following a standard protocol [
29], with two pulses of 6 m/s for 30 s in a FAST- PREP24 (Biomedicals, Irving, CA, USA). Extracted carotenoids were concentrated and measured as described [
27].
2.9. Statistical Analysis
Unpaired and paired
t Student tests were used to analyze differences in carotenoid accumulation and gene expression with GraphPad Prism 8 application (
https://graphpad.com).
Values of p lower than 0.05 were considered significantly different. Levels of significance are indicated with asterisks: * indicates p < 0.05; ** indicates p < 0.01; *** p < 0.001 and **** p < 0.0001.
4. Discussion
The Tet-on system is a highly versatile tool to control gene expression that has been successfully used in different filamentous fungi, such as
A. niger,
A. fumigatus,
A. terreus [
21,
22,
30,
31,
32,
33], and
F. fujikuroi [
24], among others. In the latter, a gene for transcription factor TF22 from a silent cluster was expressed at different levels using the bacterial–fungal hybrid promoter
tetO7::P
poliC system, and in TET::
TF22, upregulation of three genes of the cluster for trichosetin was achieved [
24].
In our case, the expression system was tuned up in TET
luc transformants with the reporter gene
mluc under the control of the
tetO7::Pmin promoter. TET
luc transformants have the advantage that luciferase activity is easy to measure, and they produced high luminescence in just few hours after the induction with Dox (
Figure 2A). The rapid and strong induction of
mluc in these strains is comparable to that formerly observed in similar transformants of
A. niger [
20]. The luciferase activity had a significant induction with 20 µg/mL Dox, and it could predictably keep increasing with higher concentrations. In fact, in other studies, high inductions were achieved with 50 µg/mL Dox; however, at that concentration, the growth of wild-type
F. fujikuroi was appreciably inhibited on complete agar medium [
24]. We detected that inhibition occurred with only 20 µg/mL Dox in solid cultures on minimal medium in the
carS mutant SG39 and its derived transformant, but not in the wild type or its derived strain (
Figure 3 and
Figure 5). However, in microtiter plates, the inhibition with 20 µg/mL was not statistically significant, although the luciferase activity was outstanding. Moreover, optimization of culture conditions, such as aeration, culture composition, and illumination, reduces the toxicity caused by this tetracycline-derived antibiotic, which has been described as a selective inhibitor of mitochondrial protein translation [
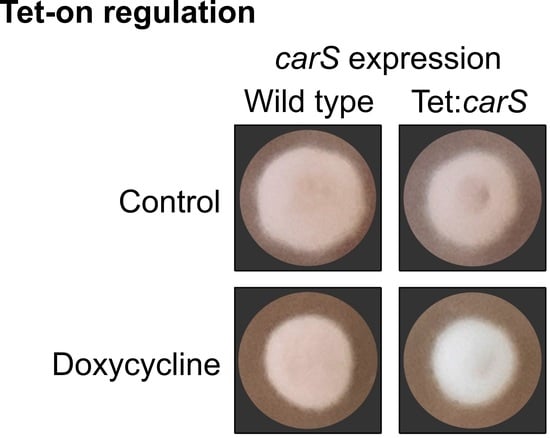
34]. For this reason, we recommend checking the effect of this antibiotic on the growth of the organism object of experimentation before using this Tet-on system.
Tet-on is an experimentally convenient inducible expression system that has been used for different purposes in different biological contexts. In has been used to express a polycistronic mRNA in
A. niger [
35] and to control gene expression by blue light- and Dox-dependent manner in mammalian cells [
36]. One of the advantages of the Tet-on approach is that it allows studying phenotypes that could not be observed when the high expression of a regulator, an activator, or a repressor affects the viability of the transformant due to the production of secondary metabolites. We predict that this system will continually improve and will be used for new applications in the future to study gene regulation and to control the expression of genes for the production of metabolites of interest.
Since the identification of the
carS gene, resulting from the analysis of mutations responsible of the deep orange pigmentation of a class of
F. fujikuroi mutants [
9], several studies have been performed to understand its role in the regulation of carotenoid biosynthesis [
7]. This pathway is mainly regulated by light and to a minor extent by other environmental signals, such as nitrogen availability. The major regulatory protein involved in the regulation by light is WcoA, coding for a photoreceptor of the white-collar family [
37]. However, the pathway is downregulated by the CarS protein, as indicates the strong increase in the mRNA levels of the
car genes in the
carS mutants [
7]. Such mutants, however, still respond to light, and the possible involvement of CarS in the regulation mediated by WcoA remains to be investigated.
RNA-seq data on the effects of light and
carS mutation in
F. fujikuroi showed a high overlap between the genes regulated by light and those differentially expressed in the
carS mutant, suggesting regulatory connections [
18]. This indicates that CarS plays a role as a modulator of many light-regulated genes, but it does not imply a direct participation of CarS in the control by light. In this study, the increase in
carS mRNA using either a Dox-inducible Tet-on promoter or a constitutive P
gpdA promoter gave albino phenotypes. The results were similar with both strategies and showed that the increased levels of
carS correlated with a complete reduction of mRNA levels of the structural genes
carRA and
carB. This is consistent with a dose-specific action of CarS, in which an excess of CarS protein results in an over-repressed carotenoid pathway. A partial repression is already operating in the wild strain, with presumable scarce CarS levels, as indicated by the low number of transcripts detected in the RNAseq studies and the high expression of the genes of the carotenoid pathway in the absence of the functional CarS protein. A novel lncRNA gene, located upstream to
carS, may be at least partially responsible for the attenuated
carS expression [
38]. However, mRNA levels do not necessarily correlate with functional protein levels, since regulatory proteins are frequently the subject of interactions with other proteins that generate posttranslational modifications to modulate their activity or trigger their degradation. As an example, the white-collar protein WC-1, the major transcriptional activator of carotenogenesis in
Neurospora crassa, is phosphorylated together with its WC-2 partner in the white-collar complex after activation by light, impairing its activating function [
39]. The protein CarS is likely to be subject to a similar regulation. While its mRNA levels are higher after illumination, the carotenoid levels do also increase, which is the opposite effect that we would expect from a higher
carS expression. In our case, however, the decreased carotenoid biosynthesis under CarS overproduction was also observed in the light.
In this study, the levels of
carS in the OE
carS strains were higher that with the Tet-on system and as a result provoked a complete repression of
carB and carotenoid synthesis, while in the TET
carS transformants, this gene was not fully repressed with the assayed Dox concentration. These results agree with other studies in which overexpression of a transcription factor with a constitutive promoter gave a higher production of trichosetin than with a Dox-inducible hybrid promoter [
24]. Our findings reinforce the idea that the physiological levels of CarS are precisely tuned to maintain the amounts of carotenoids in the dark at low levels while still allowing a sufficient induction of the synthesis under illumination. Moreover, our data are consistent with the occurrence of a posttranscriptional regulation of
carS, as indicated by the higher levels of
carS mRNA in the light when the
carS gene is overexpressed in the OE
carS strains, considering that the P
gpd promoter is not regulated by light. This increase could not to be explained by the
carS expression from the native promoter, still present in the genome for the
carS wild-type allele, because although relative
carS mRNA values are higher in the wild strain in the light compared to the dark, the increase is quantitatively much higher in the SG263 and SG264 strains. As CarS is a repressor of
car genes, the existence of a regulatory system to counteract the activating effect of light would be expectable, possibly involving a higher stability of
carS transcripts in the light, but not necessarily implying a higher availability of the active CarS protein.
The fine regulation of carotenogenesis by the CarS protein in
F. fujikuroi and its connections with the control by light is an intriguing scientific issue with potential biotechnological applications. Because of its antioxidant properties [
4], NX is an attractive product for biotechnologists, and the understanding of the molecular mechanism that controls its synthesis in this fungus may have future applications. In this respect, the understanding of the posttranscriptional mechanisms governing
carS function will be of particular interest in future research. For this, and for other purposes, the successful use of inducible expression systems, such as the Tet-on used in this work, is also very promising.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}