New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
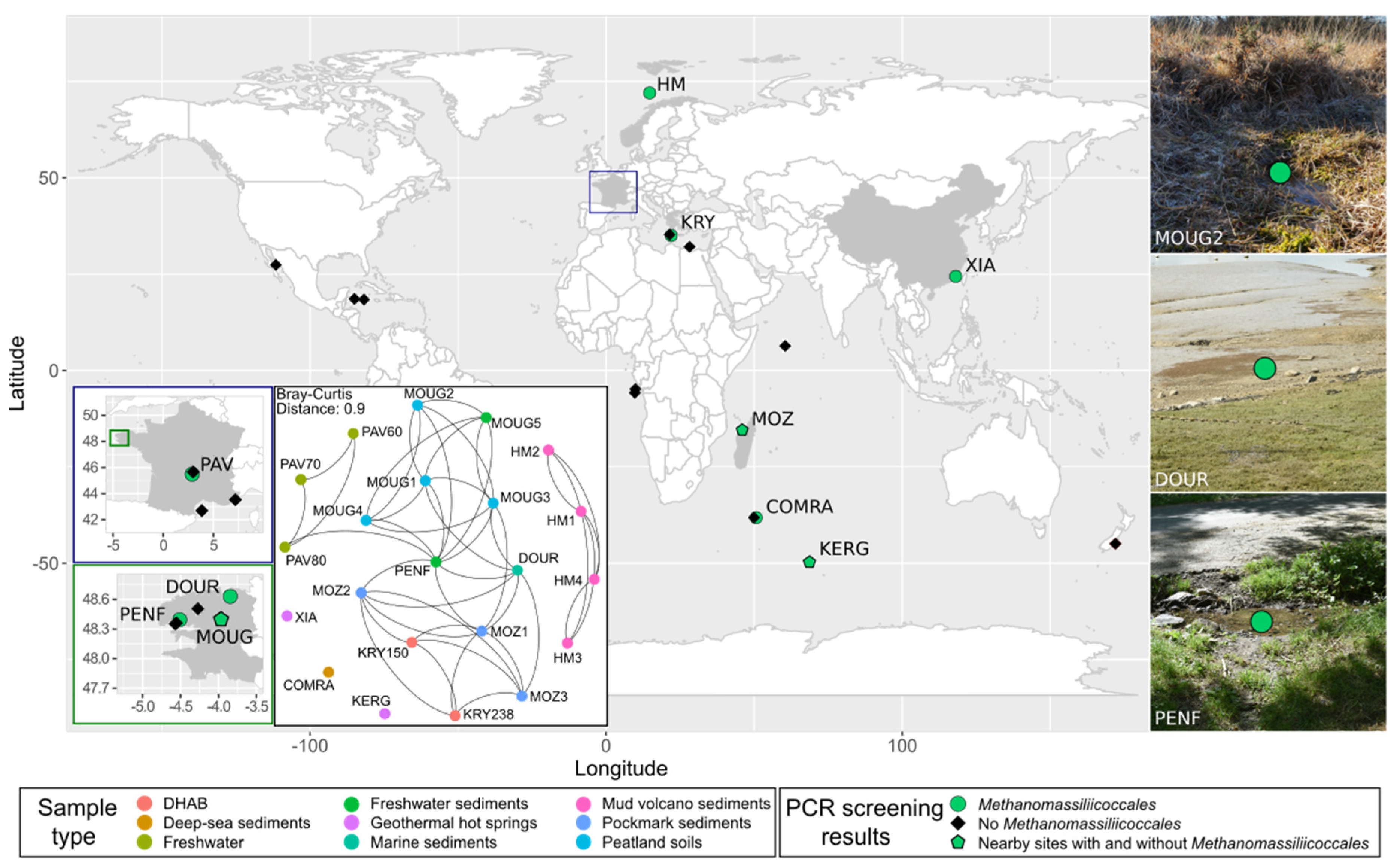
2.1. Site Description
2.2. DNA Extraction
2.3. Metabarcoding Sequencing and Sequence Analysis
2.4. Quantitative Polymerase Chain Reaction
2.5. Culture-Based Incubation Experiments
2.5.1. Medium Composition for Culture-Based Experiments with Environmental Slurries
2.5.2. Monitoring of Culture-Based Incubation Experiments
2.5.3. Quantifications of Substrates and Metabolic Products
2.6. MAG Sequencing and Annotation
2.7. Phylogenetic Position of the MAGs and Comparative Genomics
2.8. Co-Occurrence Network Analysis
2.9. Availability of Data and Materials
3. Results and Discussion
3.1. Diversity and Abundance of Methanomassiliicoccales in a Wide Range of Habitats
3.1.1. Molecular Pre-Screening of Various Environmental Samples
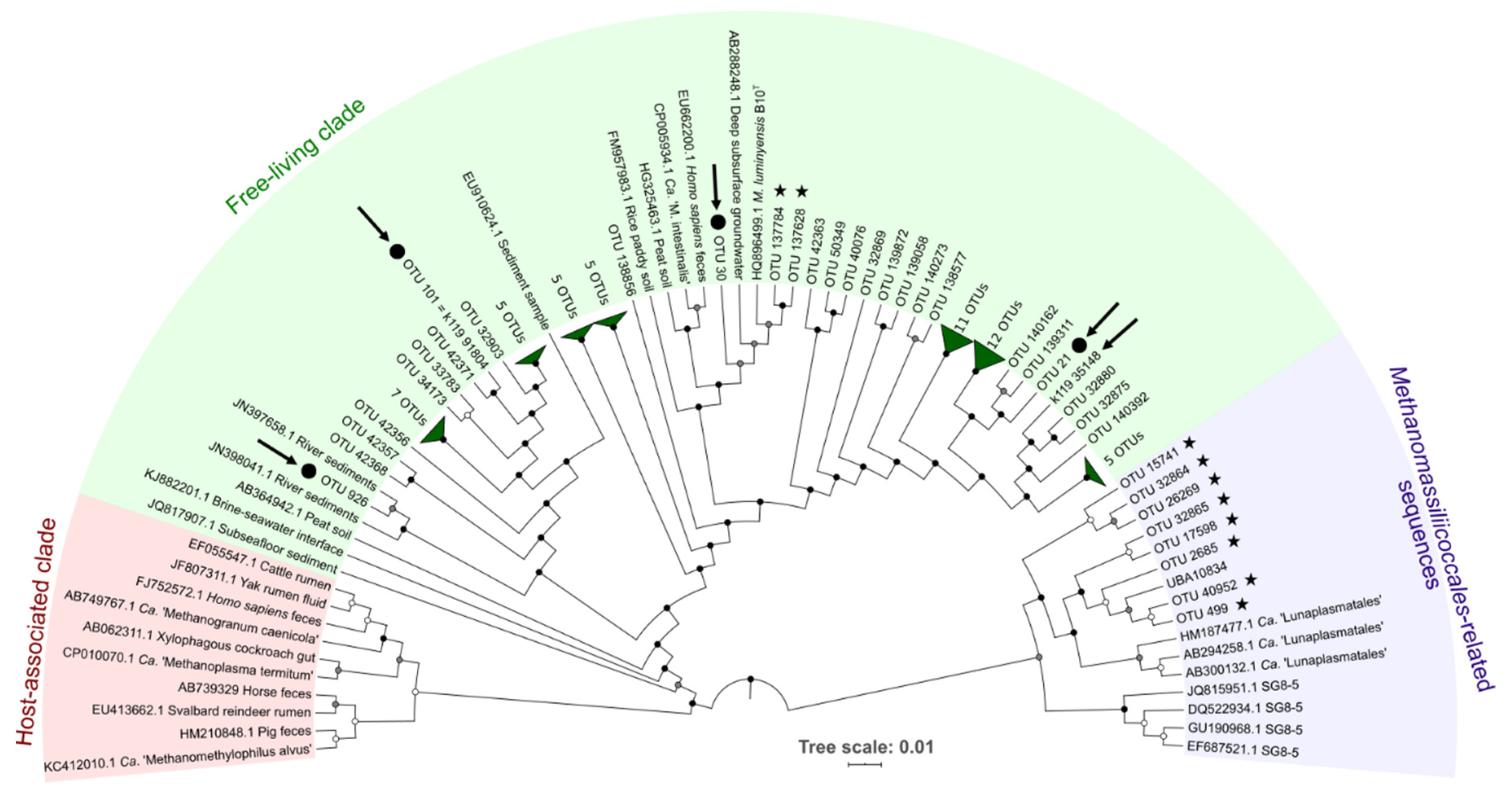
3.1.2. Diversity and Abundance of Methanomassiliicoccales
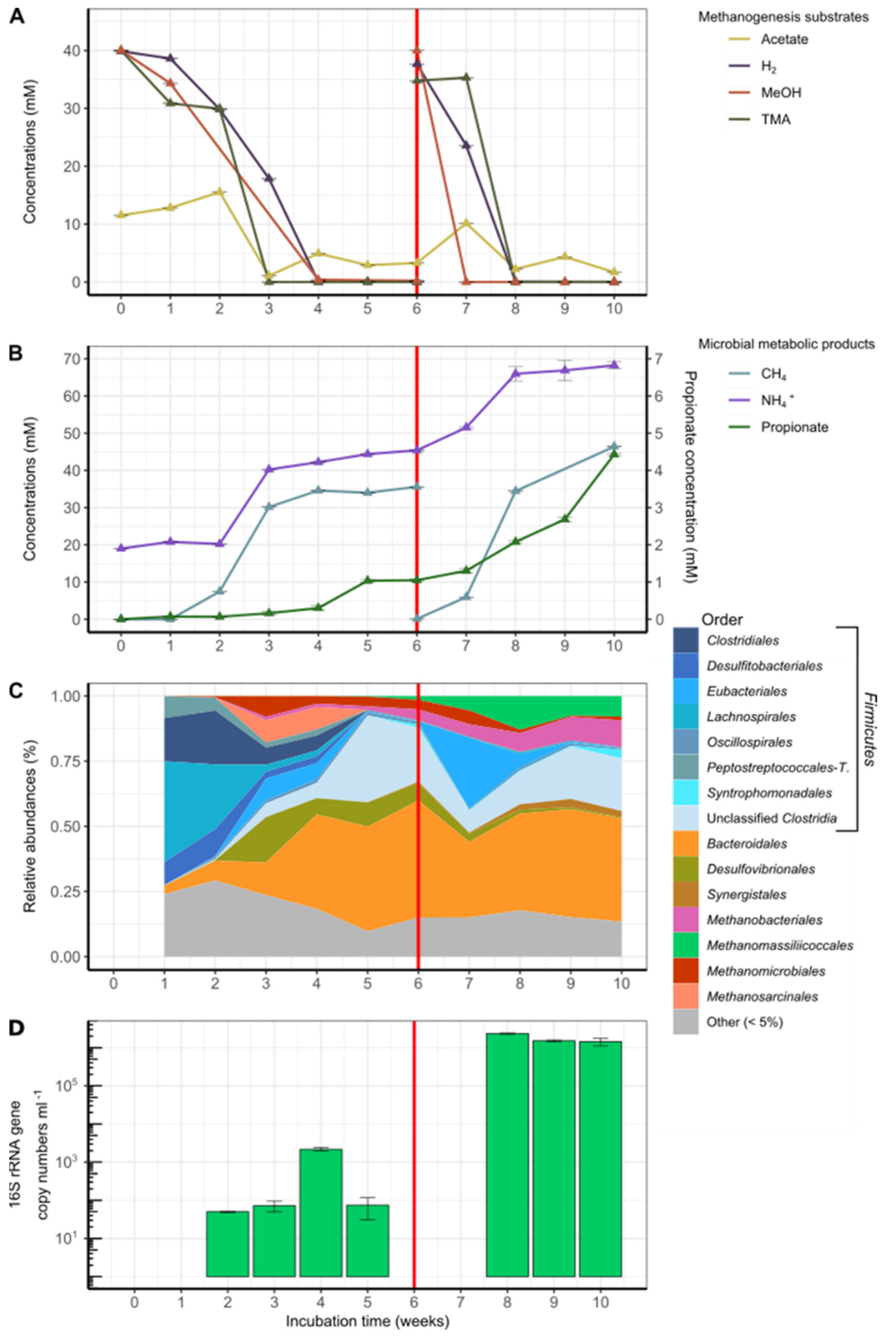
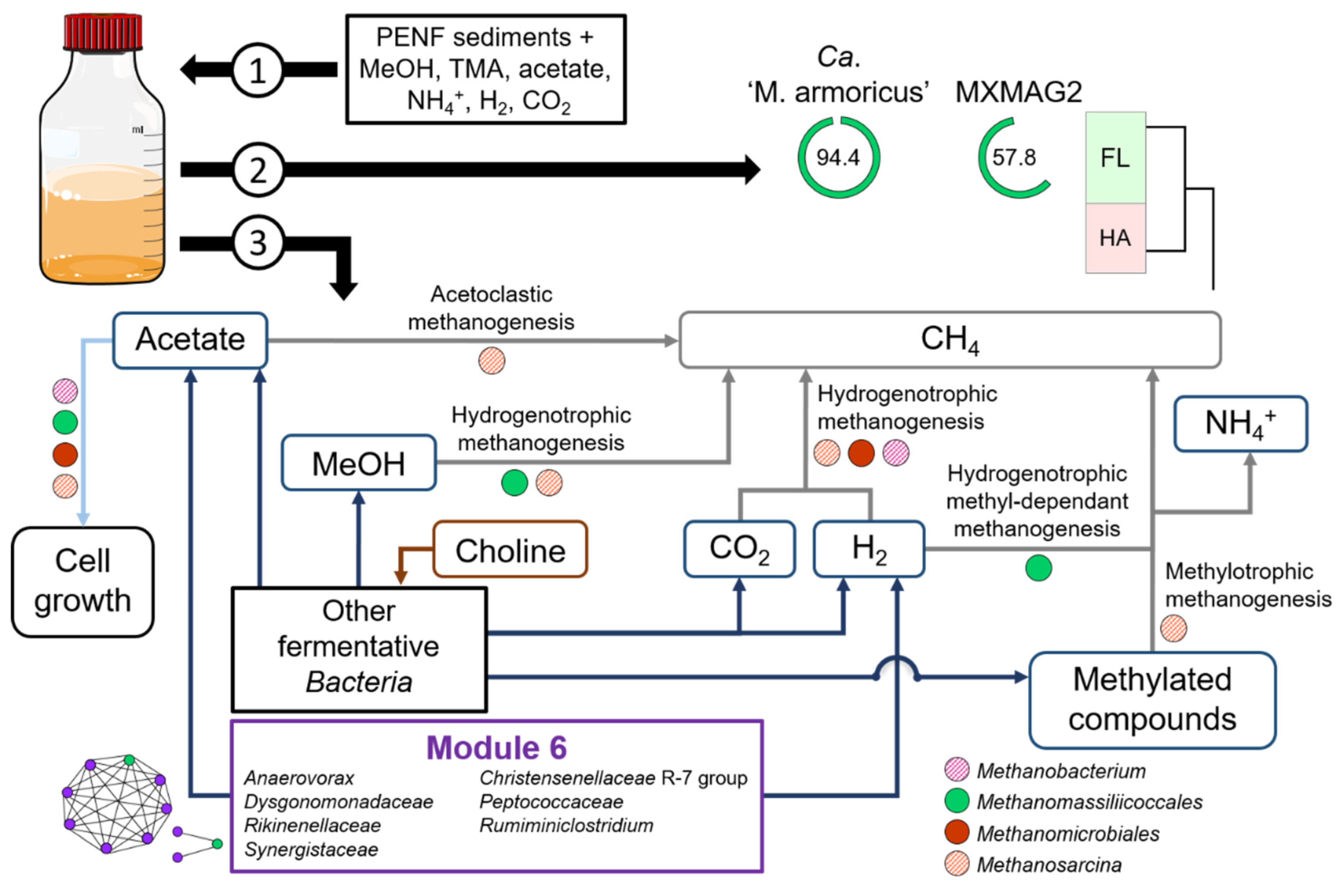
3.2. Enrichment Cultures Targeting Methanomassiliicoccales
3.3. Methanomassiliicoccales Genomic Features
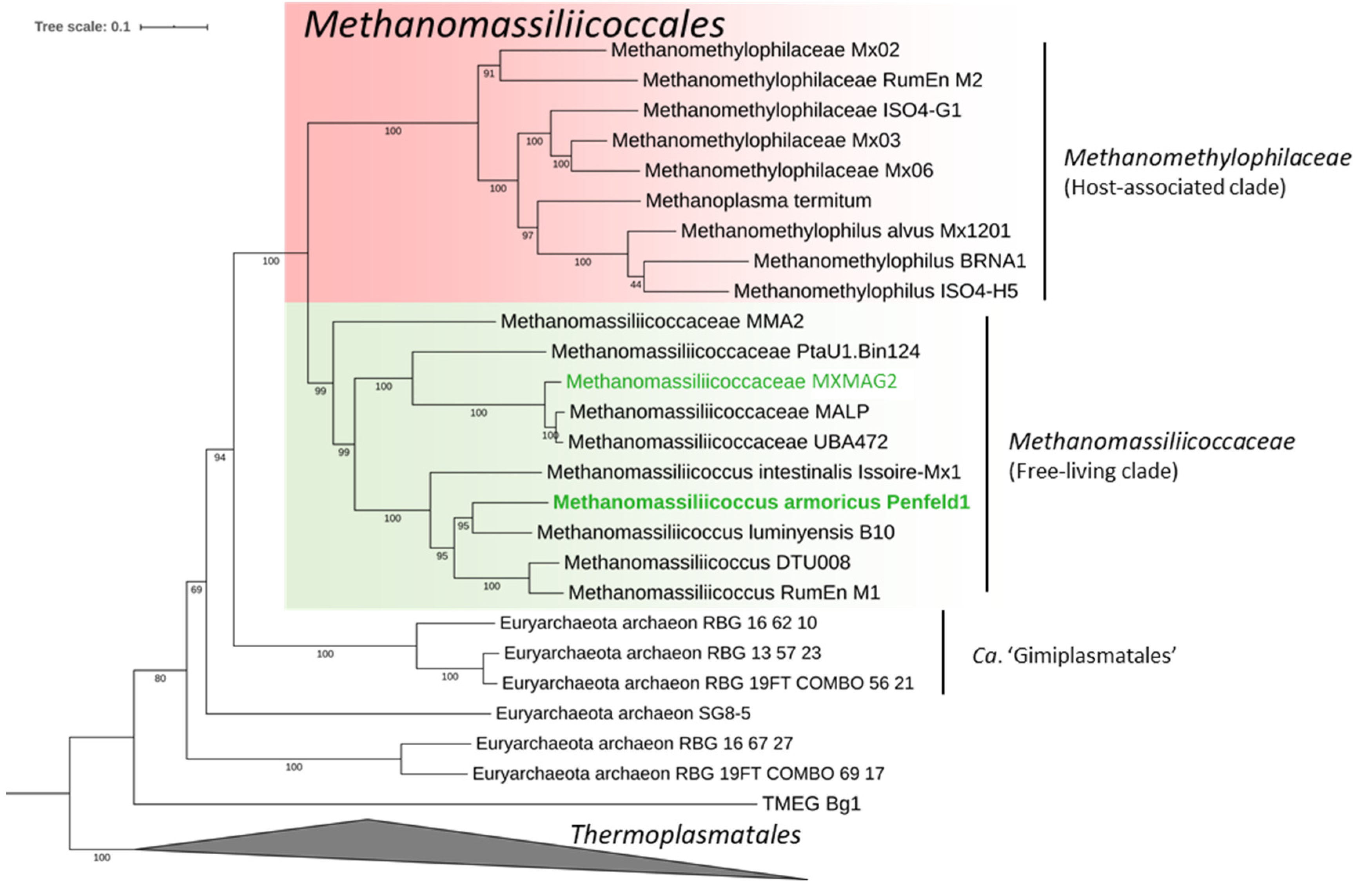
3.3.1. Taxonomic Position and General Features of the MAGs
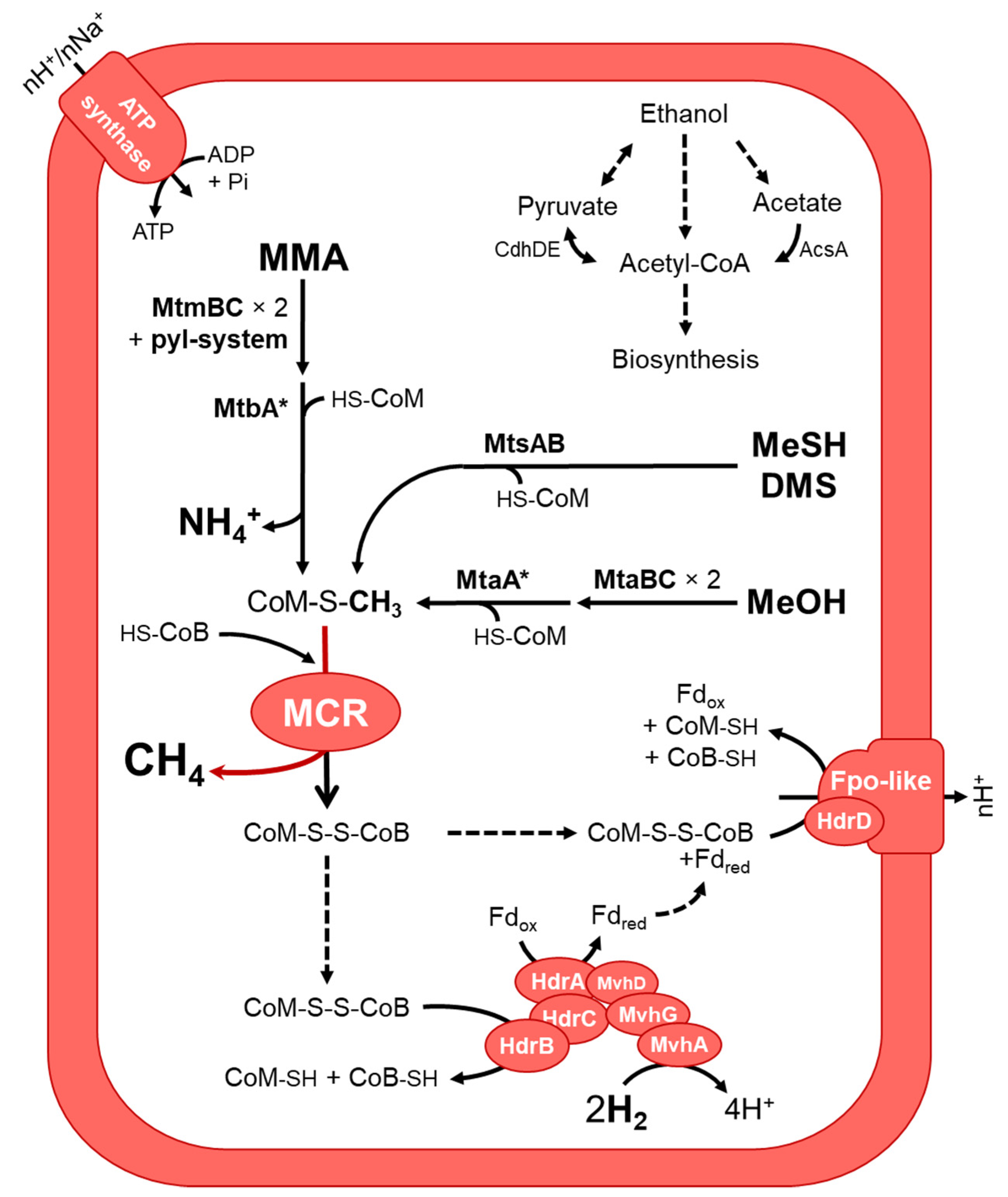
3.3.2. Putative Metabolic Pathways
3.3.3. Putative Physiological and Stress Response Functions
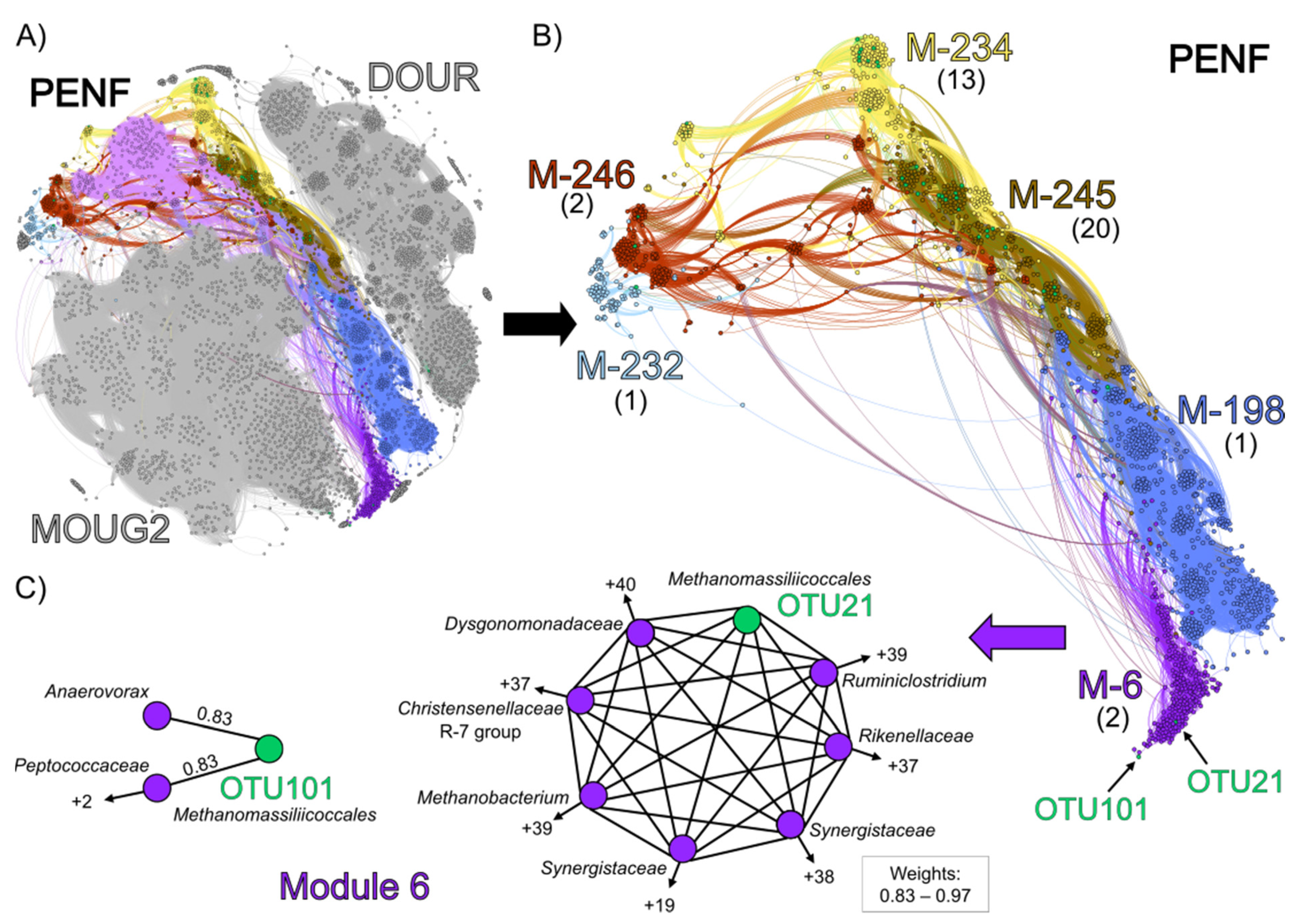
3.4. Co-Occurrence Network Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| Ala | Alanine |
| ANI | Average Nucleotide Identity |
| ANOVA | Analysis of variance |
| Arg | Arginine |
| ASCII | American Standard Code for Information Interchange |
| ATP | Adenosine triphosphate |
| Asp | Asparagine |
| BIOM | Biological Observation Matrix |
| Bp | Base pair |
| BSA | Bovine Serum Albumin Acetylated |
| Ca. | Candidatus |
| CERS | Cation Electrolytically Regenerated Suppressor |
| CDS | Coding DNA sequences |
| CSS | Cumulative Sum Scaling |
| Cys | Cystine |
| DES | Deionized Elution Buffer |
| DDH | DNA-DNA Hybridization |
| Dfast | DDBJ Fast Annotation and Submission Tool |
| DHAB | Deep-sea Hypersaline Anoxic Brine |
| DHVEG-1 | Deep-sea Hydrothermal Vent Euryarchaeota Group-1 |
| DMA | Dimethylamine |
| DMS | Dimethylsulfide |
| DNA | Desoxyribonucleic Acid |
| DSMZ | Deutsche Sammlung von Mikroorganismen und Zellkulturen |
| EB | Elution Buffer |
| FID | Flame Ionization Detector |
| GC | Gas Chromatograph |
| GGDC | Genome to Genome Distance Calculator |
| GIT | Gastro-Intestinal Tract |
| Glu | Glutamine |
| Gly | Glycine |
| GTDB-Tk | Genome Taxonomy Database Toolkit |
| HDR | HeteroDisulfide Reductase |
| His | Histidine |
| Ile | Isoleucine |
| iTOL | interactive Tree Of Life |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| Lys | Lysine |
| MAG | Metagenome-Assembled Genome |
| MaGe | MicroScope Microbial Genome Annotation and Analysis Platform |
| MBGD | Marine Benthic Group D |
| MCR | Methyl-Coenzyme M Reductase |
| MeOH | Methanol |
| MeSH | Methanethiol |
| MetaBAT | Metagenome Binning with Abundance and Tetra-nucleotide frequencies |
| MMA | Monomethylamine |
| MUSCLE | Multiple Sequence Comparison by Log- Expectation |
| NCBI | National Center for Biotechnology Information |
| OTU | Operational Taxonomic Unit |
| PAMELA | Passive Margins Exploration Laboratories |
| PCI | Phenol Chloroform Isoamyl alcohol |
| PCR | Polymerase Chain Reaction |
| Pyl | Pyrrolysine |
| Prokka | rapid prokaryotic genome annotation |
| qPCR | Quantitative Polymerase Chain Reaction |
| Quast | Quality Assessment Tool |
| RAST | Rapid Annotations using Subsystems Technology |
| RDP | Ribosomal Database Project |
| rRNA | ribosomal Ribonucleic Acid |
| SDS | Sodium Dodecyl Sulfate |
| SSU | Small Subunit |
| TE | Tris-Ethylene diamine tetra acetic acid |
| Thr | Threonine |
| TMA | Trimethylamine |
| tRNA | transfer RNA |
| Trp | Tryptophane |
| UniProtKB | Universal Protein resource Knowledgebase |
| Val | Valine |
References
- Kirschke, S.; Bousquet, P.; Ciais, P.; Saunois, M.; Canadell, J.G.; Dlugokencky, E.J.; Bergamaschi, P.; Bergmann, D.; Blake, D.R.; Bruhwiler, L.; et al. Three decades of global methane sources and sinks. Nat. Geosci. 2013, 6, 813–823. [Google Scholar] [CrossRef]
- Chaban, B.; Ng, S.Y.M.; Jarrell, K.F. Archaeal habitats—From the extreme to the ordinary. Can. J. Microbiol. 2006, 52, 73–116. [Google Scholar] [CrossRef] [PubMed]
- Zinder, S.H. Physiological Ecology of Methanogens. In Methanogenesis: Ecology, Physiology, Biochemistry & Genetics; Ferry, J.G., Ed.; Chapman & Hall: New York, NY, USA, 1993; ISBN 978-1-4615-2391-8. [Google Scholar] [CrossRef]
- Garcia, J.-L.; Patel, B.K.C.; Ollivier, B. Taxonomic, Phylogenetic, and Ecological Diversity of Methanogenic Archaea. Anaerobe 2000, 6, 205–226. [Google Scholar] [CrossRef] [PubMed]
- Parkes, R.J.; Cragg, B.; Roussel, E.; Webster, G.; Weightman, A.; Sass, H. A review of prokaryotic populations and processes in sub-seafloor sediments, including biosphere: Geosphere interactions. Marine Geol. 2014, 352, 409–425. [Google Scholar] [CrossRef]
- Evans, P.N.; Parks, D.H.; Chadwick, G.L.; Robbins, S.J.; Orphan, V.J.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef]
- Mayumi, D.; Mochimaru, H.; Tamaki, H.; Yamamoto, K.; Yoshioka, H.; Suzuki, Y.; Kamagata, Y.; Sakata, S. Methane production from coal by a single methanogen. Science 2016, 354, 222–225. [Google Scholar] [CrossRef]
- Borrel, G.; Adam, P.S.; McKay, L.J.; Chen, L.-X.; Sierra-García, I.N.; Sieber, C.M.K.; Letourneur, Q.; Ghozlane, A.; Andersen, G.L.; Li, W.-J.; et al. Wide diversity of methane and short-chain alkane metabolisms in uncultured archaea. Nat. Microbiol. 2019, 4, 603–613. [Google Scholar] [CrossRef]
- Borrel, G.; Brugère, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636. [Google Scholar] [CrossRef]
- Nobu, M.K.; Narihiro, T.; Kuroda, K.; Mei, R.; Liu, W.-T. Chasing the elusive Euryarchaeota class WSA2: Genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 2016, 10, 2478–2487. [Google Scholar] [CrossRef]
- Vanwonterghem, I.; Evans, P.N.; Parks, D.H.; Jensen, P.D.; Woodcroft, B.J.; Hugenholtz, P.; Tyson, G.W. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 2016, 1, 16170. [Google Scholar] [CrossRef]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Adam, P.S.; Gribaldo, S. Methanogenesis and the Wood—Ljungdahl Pathway: An Ancient, Versatile, and Fragile Association. Genome Biol. Evol. 2016, 8, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Dridi, B.; Fardeau, M.-L.; Ollivier, B.; Raoult, D.; Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Micr. 2012, 62, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; Gaci, N.; Peyret, P.; O’Toole, P.W.; Gribaldo, S.; Brugère, J.-F. Unique Characteristics of the Pyrrolysine System in the 7th Order of Methanogens: Implications for the Evolution of a Genetic Code Expansion Cassette. Archaea 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Kröninger, L.; Berger, S.; Welte, C.; Deppenmeier, U. Evidence for the involvement of two heterodisulfide reductases in the energy-conserving system of Methanomassiliicoccus luminyensis. FEBS J. 2016, 283, 472–483. [Google Scholar] [CrossRef]
- Borrel, G.; Harris, H.M.B.; Tottey, W.; Mihajlovski, A.; Parisot, N.; Peyretaillade, E.; Peyret, P.; Gribaldo, S.; O’Toole, P.W.; Brugère, J.-F. Genome Sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a Methanogenic Archaeon from the Human Gut Belonging to a Seventh Order of Methanogens. J. Bacteriol. 2012, 194, 6944–6945. [Google Scholar] [CrossRef]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugere, J.-F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef]
- Iino, T.; Tamaki, H.; Tamazawa, S.; Ueno, Y.; Ohkuma, M.; Suzuki, K.; Igarashi, Y.; Haruta, S. Candidatus Methanogranum caenicola: A Novel Methanogen from the Anaerobic Digested Sludge, and Proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a Methanogenic Lineage of the Class Thermoplasmata. Microbes Environ. 2013, 28. [Google Scholar] [CrossRef]
- Paul, K.; Nonoh, J.O.; Mikulski, L.; Brune, A. “Methanoplasmatales,” Thermoplasmatales-Related Archaea in Termite Guts and Other Environments, Are the Seventh Order of Methanogens. Appl. Environ. Microbiol. 2012, 78, 8245–8253. [Google Scholar] [CrossRef]
- Borrel, G.; O’Toole, P.W.; Harris, H.M.B.; Peyret, P.; Brugère, J.-F.; Gribaldo, S. Phylogenomic Data Support a Seventh Order of Methylotrophic Methanogens and Provide Insights into the Evolution of Methanogenesis. Genome Biol. Evol. 2013, 5, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Söllinger, A.; Schwab, C.; Weinmaier, T.; Loy, A.; Tveit, A.T.; Schleper, C.; Urich, T. Phylogenetic and genomic analysis of Methanomassiliicoccales in wetlands and animal intestinal tracts reveals clade-specific habitat preferences. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed]
- Borrel, G.; McCann, A.; Deane, J.; Neto, M.C.; Lynch, D.B.; Brugère, J.-F.; O’Toole, P.W. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. ISME J. 2017, 11, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- De la Cuesta-Zuluaga, J.; Spector, T.D.; Youngblut, N.D.; Ley, R.E. Genomic insights into adaptations of TMA-utilizing methanogens to diverse habitats including the human gut. bioRxiv 2020. Unpublished work. [Google Scholar] [CrossRef]
- Lang, K.; Schuldes, J.; Klingl, A.; Poehlein, A.; Daniel, R.; Brune, A. New Mode of Energy Metabolism in the Seventh Order of Methanogens as Revealed by Comparative Genome Analysis of “Candidatus Methanoplasma termitum”. Appl. Environ. Microbiol. 2015, 81, 1338–1352. [Google Scholar] [CrossRef]
- Li, Y.; Leahy, S.C.; Jeyanathan, J.; Henderson, G.; Cox, F.; Altermann, E.; Kelly, W.J.; Lambie, S.C.; Janssen, P.H.; Rakonjac, J.; et al. The complete genome sequence of the methanogenic archaeon ISO4-H5 provides insights into the methylotrophic lifestyle of a ruminal representative of the Methanomassiliicoccales. Stand. Genomic Sci. 2016, 11, 59. [Google Scholar] [CrossRef]
- Mihajlovski, A.; Doré, J.; Levenez, F.; Alric, M.; Brugère, J.-F. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ. Microbiol. Rep. 2010, 2, 272–280. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef]
- Vanderhaeghen, S.; Lacroix, C.; Schwab, C. Methanogen communities in stools of humans of different age and health status and co-occurrence with bacteria. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef]
- De León, K.B.; Gerlach, R.; Peyton, B.M.; Fields, M.W. Archaeal and bacterial communities in three alkaline hot springs in Heart Lake Geyser Basin, Yellowstone National Park. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Cao, H.; Han, P.; Gu, J.-D. Analysis of methane-producing and metabolizing archaeal and bacterial communities in sediments of the northern South China Sea and coastal Mai Po Nature Reserve revealed by PCR amplification of mcrA and pmoA genes. Front. Microbiol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Guan, W.; Chen, H.; Liao, B.; Hu, J.; Peng, C.; Rui, J.; Tian, J.; Zhu, D.; He, Y. Archaeal communities in the sediments of different mangrove stands at Dongzhaigang, China. J. Soils Sediments 2016, 16, 1995–2004. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Pan, J.; Liu, Y.; Duan, C.-H.; Li, M. Genomic and transcriptomic insights into methanogenesis potential of novel methanogens from mangrove sediments. Microbiome 2020, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2016, 1. [Google Scholar] [CrossRef]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2018, 34, 1287–1294. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- Paulson, J.N.; Pop, M.; Bravo, H.C. metagenomeSeq: Statistical Analysis for Sparse High-Throughput Sequencing. Available online: http://cbcb.umd.edu/software/metagenomeSeq (accessed on 20 November 2020.).
- Kemnitz, D.; Kolb, S.; Conrad, R. Phenotypic characterization of Rice Cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ. Microbiol. 2005, 7, 553–565. [Google Scholar] [CrossRef]
- Kröninger, L.; Gottschling, J.; Deppenmeier, U. Growth Characteristics of Methanomassiliicoccus luminyensis and expression of methyltransferase encoding genes. Archaea 2017, 2756573. [Google Scholar] [CrossRef] [PubMed]
- Widdel, F.; Kohring, G.-W.; Mayer, F. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. Arch. Microbiol. 1983, 134, 286–294. [Google Scholar] [CrossRef]
- Yakimov, M.M.; Cono, V.L.; Spada, G.L.; Bortoluzzi, G.; Messina, E.; Smedile, F.; Arcadi, E.; Borghini, M.; Ferrer, M.; Schmitt-Kopplin, P.; et al. Microbial community of the deep-sea brine Lake Kryos seawater—Brine interface is active below the chaotropicity limit of life as revealed by recovery of mRNA. Environ. Microbiol. 2015, 17, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.; Rinna, J.; Roussel, E.G.; Fry, J.C.; Weightman, A.J.; Parkes, R.J. Prokaryotic functional diversity in different biogeochemical depth zones in tidal sediments of the Severn Estuary, UK, revealed by stable-isotope probing. FEMS Microbiol. Ecol. 2010, 72, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, A.; Brisse, S. AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Crusoe, M.R.; Alameldin, H.F.; Awad, S.; Boucher, E.; Caldwell, A.; Cartwright, R.; Charbonneau, A.; Constantinides, B.; Edvenson, G.; Fay, S.; et al. The khmer software package: Enabling efficient nucleotide sequence analysis. F1000Research 2015, 4, 900. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015, 3, e1165. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Vallenet, D.; Calteau, A.; Cruveiller, S.; Gachet, M.; Lajus, A.; Josso, A.; Mercier, J.; Renaux, A.; Rollin, J.; Rouy, Z.; et al. MicroScope in 2017: An expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 2017, 45, D517–D528. [Google Scholar] [CrossRef]
- Tanizawa, Y.; Fujisawa, T.; Nakamura, Y. DFAST: A flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 2018, 34, 1037–1039. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Micr. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Aires, T.; Moalic, Y.; Serrao, E.A.; Arnaud-Haond, S. Hologenome theory supported by cooccurrence networks of species-specific bacterial communities in siphonous algae (Caulerpa). FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef]
- Moalic, Y.; Desbruyères, D.; Duarte, C.M.; Rozenfeld, A.F.; Bachraty, C.; Arnaud-Haond, S. Biogeography Revisited with Network Theory: Retracing the History of Hydrothermal Vent Communities. Syst. Biol. 2012, 61, 127. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the 3rd International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009; Volume 8, pp. 361–362. [Google Scholar]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Lovley, D.R.; Klug, M.J. Methanogenesis from Methanol and Methylamines and Acetogenesis from Hydrogen and Carbon Dioxide in the Sediments of a Eutrophic Lake. Appl. Environ. Microbiol. 1983, 45, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- King, G.M. Metabolism of Trimethylamine, Choline, and Glycine Betaine by Sulfate-Reducing and Methanogenic Bacteria in Marine Sediments. Appl. Environ. Microbiol. 1984, 48, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Formation and breakdown of glycine betaine and trimethylamine in hypersaline environments. Antonie van Leeuwenhoek 1990, 58, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Lomans, B.P.; Smolders, A.; Intven, L.M.; Pol, A.; Op, D.; Drift, C.V.D. Formation of dimethyl sulfide and methanethiol in anoxic freshwater sediments. Appl. Environ. Microbiol. 1997, 63, 4741–4747. [Google Scholar] [CrossRef] [PubMed]
- Hanna, E.; Keller, J.K.; Chang, D.; de Bruyn, W.; Zalman, C. The potential importance of methylated substrates in methane production within three northern Minnesota peatlands. Soil Biol. Biochem. 2020, 150, 107957. [Google Scholar] [CrossRef]
- Watkins, A.J.; Roussel, E.G.; Webster, G.; Parkes, R.J.; Sass, H. Choline and N,N-Dimethylethanolamine as Direct Substrates for Methanogens. Appl. Environ. Microbiol. 2012, 78, 8298–8303. [Google Scholar] [CrossRef] [PubMed]
- Merkel, A.Y.; Podosokorskaya, O.A.; Chernyh, N.A.; Bonch-Osmolovskaya, E.A. Occurrence, diversity, and abundance of methanogenic archaea in terrestrial hot springs of Kamchatka and Saõ Miguel Island. Microbiology 2015, 84, 577–583. [Google Scholar] [CrossRef]
- Merkel, A.Y.; Podosokorskaya, O.A.; Sokolova, T.G.; Bonch-Osmolovskaya, E.A. Diversity of methanogenic archaea from the 2012 terrestrial hot spring (Valley of Geysers, Kamchatka). Microbiology 2016, 85, 342–349. [Google Scholar] [CrossRef]
- Grodnitskaya, I.D.; Trusova, M.Y.; Syrtsov, S.N.; Koroban, N.V. Structure of microbial communities of peat soils in two bogs in Siberian tundra and forest zones. Microbiology 2018, 87, 89–102. [Google Scholar] [CrossRef]
- Becker, K.W.; Elling, F.J.; Yoshinaga, M.Y.; Söllinger, A.; Urich, T.; Hinrichs, K.-U. Unusual Butane- and Pentanetriol-Based Tetraether Lipids in Methanomassiliicoccus luminyensis, a Representative of the Seventh Order of Methanogens. Appl. Environ. Microbiol. 2016, 82, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Biderre-Petit, C.; Jézéquel, D.; Dugat-Bony, E.; Lopes, F.; Kuever, J.; Borrel, G.; Viollier, E.; Fonty, G.; Peyret, P. Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol. Ecol. 2011, 77, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mi, T.; Liu, Y.; Li, S.; Zhen, Y. Microbial Community Composition and Function in Sediments from the Pearl River Mouth Basin. J. Ocean Univ. China 2020, 19, 941–953. [Google Scholar] [CrossRef]
- Borrel, G.; Parisot, N.; Harris, H.M.; Peyretaillade, E.; Gaci, N.; Tottey, W.; Bardot, O.; Raymann, K.; Gribaldo, S.; Peyret, P.; et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genom. 2014, 15, 679. [Google Scholar] [CrossRef] [PubMed]
- Zinke, L.A.; Evans, P.N.; Schroeder, A.L.; Parks, D.H.; Varner, R.K.; Rich, V.I.; Tyson, G.W.; Emerson, J.B. Evidence for non-methanogenic metabolisms in globally distributed archaeal clades basal to the Methanomassiliicoccales. Environ. Microbiol. 2020. [Google Scholar] [CrossRef]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, L.; Jin, W.; Meng, Z.; Cheng, Y.; Wang, J.; Zhu, W. Methane Emission, Rumen Fermentation, and Microbial Community Response to a Nitrooxy Compound in Low-Quality Forage Fed Hu Sheep. Curr. Microbiol. 2019, 76, 435–441. [Google Scholar] [CrossRef]
- Azizi, A.; Kim, W.; Lee, J.H. Comparison of microbial communities during the anaerobic digestion of Gracilaria under mesophilic and thermophilic conditions. World J. Microbiol. Biotechnol. 2016, 32, 158. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.D.; Papiska, H.R.; Campbell, L.L. Choline fermentation by Desulfovibrio desulfuricans. J. Bacteriol. 1962, 84, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Xu, C.-Y.; Jian, J.-S.; He, W.-X.; Hou, L.; Geng, Z.-C. Seasonal dynamics of bacterial communities in a Betula albosinensis forest. Eur. J. Soil Sci. 2018, 69, 666–674. [Google Scholar] [CrossRef]
- Bhatnagar, L.; Jain, M.K.; Aubert, J.-P.; Zeikus, J.G. Comparison of Assimilatory Organic Nitrogen, Sulfur, and Carbon Sources for Growth of Methanobacterium Species. Appl. Environ. Microbiol. 1984, 48, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Evol. Microbiol. 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Krzycki, J.A. Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases. Curr. Opin. Chem. Biol. 2004, 8, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Kröninger, L.; Steiniger, F.; Berger, S.; Kraus, S.; Welte, C.U.; Deppenmeier, U. Energy conservation in the gut microbe Methanomassiliicoccus luminyensis is based on membrane-bound ferredoxin oxidation coupled to heterodisulfide reduction. FEBS J. 2019, 286, 3831–3843. [Google Scholar] [CrossRef] [PubMed]
- Nicolaus, B.; Gambacorta, A.; Basso, A.L.; Riccio, R.; De Rosa, M.; Grant, W.D. Trehalose in Archaebacteria. Syst. Appl. Microbiol. 1988, 10, 215–217. [Google Scholar] [CrossRef]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17R–27R. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.E.; Xu, H.; White, R.H. Methanococcus jannaschii Uses a Pyruvoyl-Dependent Arginine Decarboxylase in Polyamine Biosynthesis. J. Biol. Chem. 2002, 277, 23500–23507. [Google Scholar] [CrossRef]
- Tolbert, W.D.; Graham, D.E.; White, R.H.; Ealick, S.E. Pyruvoyl-Dependent Arginine Decarboxylase from Methanococcus jannaschii: Crystal Structures of the Self-Cleaved and S53A Proenzyme Forms. Structure 2003, 11, 285–294. [Google Scholar] [CrossRef][Green Version]
- Richard, H.T.; Foster, J.W. Acid resistance in Escherichia coli. Adv. Appl. Microbiol. 2003, 52, 167–186. [Google Scholar]
- Sugiyama, Y.; Nakamura, A.; Matsumoto, M.; Kanbe, A.; Sakanaka, M.; Higashi, K.; Igarashi, K.; Katayama, T.; Suzuki, H.; Kurihara, S. A Novel Putrescine Exporter SapBCDF of Escherichia coli. J. Biol. Chem. 2016, 291, 26343–26351. [Google Scholar] [CrossRef]
- Scherer, P.; Kneifel, H. Distribution of polyamines in methanogenic bacteria. J. Bacteriol. 1983, 154, 1315–1322. [Google Scholar] [CrossRef]
- Michael, A.J. Polyamine function in archaea and bacteria. J. Biol. Chem. 2018, 293, 18693–18701. [Google Scholar] [CrossRef]
- Sierra-Alvarez, R.; Cortinas, I.; Yenal, U.; Field, J.A. Methanogenic Inhibition by Arsenic Compounds. Appl. Environ. Microbiol. 2004, 70, 5688–5691. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rosen, B. Biosensors for Inorganic and Organic Arsenicals. Biosensors 2014, 4, 494–512. [Google Scholar] [CrossRef] [PubMed]
- Field, J.A.; Sierra-Alvarez, R.; Cortinas, I.; Feijoo, G.; Moreira, M.T.; Kopplin, M.; Gandolfi, A.J. Facile reduction of arsenate in methanogenic sludge. Biodegradation 2004, 15, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Rensing, C.; Rosen, B.P. Toxicity: Resistance Pathways for Metalloids and Toxic Metals. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Chichester, WS, UK, 2013; pp. 1–13. ISBN 978-1-119-95143-8. [Google Scholar] [CrossRef]
- Hu, W.; Pan, J.; Wang, B.; Guo, J.; Li, M.; Xu, M. Metagenomic insights into the metabolism and evolution of a new Thermoplasmata order (Candidatus Gimiplasmatales). Environ. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bentley, R.; Chasteen, T.G. Microbial Methylation of Metalloids: Arsenic, Antimony, and Bismuth. Microbiol. Mol. Biol. Rev. 2002, 66, 250–271. [Google Scholar] [CrossRef] [PubMed]
- Podar, M.; Gilmour, C.C.; Brandt, C.C.; Soren, A.; Brown, S.D.; Crable, B.R.; Palumbo, A.V.; Somenahally, A.C.; Elias, D.A. Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci. Adv. 2015, 1, e1500675. [Google Scholar] [CrossRef]
- Parks, J.M.; Johs, A.; Podar, M.; Bridou, R.; Hurt, R.A.; Smith, S.D.; Tomanicek, S.J.; Qian, Y.; Brown, S.D.; Brandt, C.C.; et al. The Genetic Basis for Bacterial Mercury Methylation. Science 2013, 339, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Du, H.; Wang, D. Mercury methylation by anaerobic microorganisms: A review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 1893–1936. [Google Scholar] [CrossRef]
- Gies, E.A.; Konwar, K.M.; Beatty, J.T.; Hallam, S.J. Illuminating Microbial Dark Matter in Meromictic Sakinaw Lake. Appl. Environ. Microbiol. 2014, 80, 6807–6818. [Google Scholar] [CrossRef]
- Kadnikov, V.V.; Mardanov, A.V.; Beletsky, A.V.; Karnachuk, O.V.; Ravin, N.V. Genome of the candidate phylum Aminicenantes bacterium from a deep subsurface thermal aquifer revealed its fermentative saccharolytic lifestyle. Extremophiles 2019, 23, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, M.; Castelle, C.J.; Probst, A.J.; Zhou, Z.; Pan, J.; Liu, Y.; Banfield, J.F.; Gu, J.-D. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 2018, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Álvaro, M.; Auffret, M.D.; Stewart, R.D.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; Wallace, R.J.; Shih, B.; Freeman, T.C.; Watson, M.; et al. Identification of Complex Rumen Microbiome Interaction Within Diverse Functional Niches as Mechanisms Affecting the Variation of Methane Emissions in Bovine. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Klose, M.; Conrad, R. Temperature effects on structure and function of the methanogenic microbial communities in two paddy soils and one desert soil. Soil Biol. Biochem. 2018, 124, 236–244. [Google Scholar] [CrossRef]
- Drake, H.L.; Gössner, A.S. Sporomusa . In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–7. ISBN 978-1-118-96060-8. [Google Scholar] [CrossRef]
- Schink, B. Anaerovorax . In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 1–3. ISBN 978-1-118-96060-8. [Google Scholar] [CrossRef]
- Zhang, S.; Chang, J.; Lin, C.; Pan, Y.; Cui, K.; Zhang, X.; Liang, P.; Huang, X. Enhancement of methanogenesis via direct interspecies electron transfer between Geobacteraceae and Methanosaetaceae conducted by granular activated carbon. Bioresour. Technol. 2017, 245, 132–137. [Google Scholar] [CrossRef]
- Snell-Castro, R.; Méndez-Acosta, H.O.; Arreola-Vargas, J.; González-Álvarez, V.; Pintado-González, M.; González-Morales, M.T.; Godon, J.J. Active prokaryotic population dynamics exhibit high correlation to reactor performance during methane production from acid hydrolysates of Agave tequilana var. azul bagasse. J. Appl. Microbiol. 2019, 126, 1618–1630. [Google Scholar] [CrossRef]
- Sakai, S.; Ehara, M.; Tseng, I.-C.; Yamaguchi, T.; Bräuer, S.L.; Cadillo-Quiroz, H.; Zinder, S.H.; Imachi, H. Methanolinea mesophila sp. nov., a hydrogenotrophic methanogen isolated from rice field soil, and proposal of the archaeal family Methanoregulaceae fam. nov. within the order Methanomicrobiales. Int. J. Syst. Evol. Microbiol. 2012, 62, 1389–1395. [Google Scholar] [CrossRef]
- Suzuki, D.; Li, Z.; Cui, X.; Zhang, C.; Katayama, A. Reclassification of Desulfobacterium anilini as Desulfatiglans anilini comb. nov. within Desulfatiglans gen. nov., and description of a 4-chlorophenol-degrading sulfate-reducing bacterium, Desulfatiglans parachlorophenolica sp. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 3081–3086. [Google Scholar] [CrossRef]
- Zinder, S.; Bräuer, S. Methanoregula . In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 1–8. ISBN 978-1-118-96060-8. [Google Scholar] [CrossRef]
- Morotomi, M.; Nagai, F.; Watanabe, Y. Description of Christensenella minuta gen. nov., sp. nov., isolated from human faeces, which forms a distinct branch in the order Clostridiales, and proposal of Christensenellaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 144–149. [Google Scholar] [CrossRef]
- Graf, J. The Family Rikenellaceae. In The Prokaryotes: Other Major Lineages of Bacteria and The Archaea; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 857–859. ISBN 978-3-642-38954-2. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Wiegel, J. Gracilibacteraceae . In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–3. ISBN 978-1-118-96060-8. [Google Scholar] [CrossRef]
- Shiratori-Takano, H.; Ueda, K. Lutispora . In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 1–6. ISBN 978-1-118-96060-8. [Google Scholar] [CrossRef]
- Murakami, T.; Segawa, T.; Takeuchi, N.; Sepúlveda, G.B.; Labarca, P.; Kohshima, S.; Hongoh, Y. Metagenomic analyses highlight the symbiotic association between the glacier stonefly Andiperla willinki and its bacterial gut community. Environ. Microbiol. 2018, 20, 4170–4183. [Google Scholar] [CrossRef]
- Lee, J.; Koo, T.; Yulisa, A.; Hwang, S. Magnetite as an enhancer in methanogenic degradation of volatile fatty acids under ammonia-stressed condition. J. Environ. Manag. 2019, 241, 418–426. [Google Scholar] [CrossRef]
- Waters, J.L.; Ley, R.E. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef]
- Müller, N.; Worm, P.; Schink, B.; Stams, A.J.M.; Plugge, C.M. Syntrophic butyrate and propionate oxidation processes: From genomes to reaction mechanisms. Environ. Microbiol. Rep. 2010, 2, 489–499. [Google Scholar] [CrossRef]
- Chojnacka, A.; Szczęsny, P.; Błaszczyk, M.K.; Zielenkiewicz, U.; Detman, A.; Salamon, A.; Sikora, A. Noteworthy Facts about a Methane-Producing Microbial Community Processing Acidic Effluent from Sugar Beet Molasses Fermentation. PLoS ONE 2015, 10, e0128008. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. The division “Synergistes”. Anaerobe 2007, 13, 99–106. [Google Scholar] [CrossRef]
- Yutin, N.; Galperin, M.Y. A genomic update on clostridial phylogeny: Gram-Negative spore formers and other misplaced clostridia. Environ. Microbiol. 2013, 15, 2631–2641. [Google Scholar] [CrossRef]
- McInerney, M.J.; Struchtemeyer, C.G.; Sieber, J.; Mouttaki, H.; Stams, A.J.M.; Schink, B.; Rohlin, L.; Gunsalus, R.P. Physiology, Ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann. NY Acad. Sci. 2008, 1125, 58–72. [Google Scholar] [CrossRef]
- Sieber, J.R.; McInerney, M.J.; Gunsalus, R.P. Genomic Insights into Syntrophy: The Paradigm for Anaerobic Metabolic Cooperation. Annu. Rev. Microbiol. 2012, 66, 429–452. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Acronym | Sample Type | Geographical Origin | Water Depth (m) | Depth Below Subsurface/Subseafloor (cm) | pH | Temperature (°C) |
|---|---|---|---|---|---|---|
| COMRA | Deep-sea sediments | South-West Indian ocean | 2267 | 0–20 | 7.5 | – |
| DOUR | Coastal sediments | France (Bay of Morlaix, France) | 0 | 5–10 | 7.25 | 8 |
| HM1 | Mud from a mud volcano | Barents sea, Norway | 1285 | 0–1 | – | – |
| HM2 | Mud from a mud volcano | Barents sea, Norway | 1285 | 1–6 | – | – |
| HM3 | Mud from a mud volcano | Barents sea, Norway | 1285 | 6–11 | – | – |
| HM4 | Mud from a mud volcano | Barents sea, Norway | 1282 | 1–6 | – | – |
| KERG | Water from a geothermal hot spring | Kerguelen Island, Indian Ocean (French Southern and Antarctic lands) | – | – | 7.0 | 50 |
| KRY150 | Deep-Sea Hypersaline Anoxic Basin | Mediterranean sea (Kryos basin) | ~3338 | – | 7.5 | 15 |
| KRY238 | Deep-Sea Hypersaline Anoxic Basin | Mediterranean sea (Kryos basin) | ~3338 | – | 6.5 | 15 |
| MOUG1 | Peatland soil | Peatland in France (Commana, France) | – | 0–10 | 6.4 | 4 |
| MOUG2 | Peatland soil | Peatland in France (Commana, France) | – | – | 4.78 | 2 |
| MOUG3 | Peatland soil | Peatland in France (Commana, France) | – | 0–10 | 5.22 | 6 |
| MOUG4 | Peatland soil | Peatland in France (Commana, France) | – | 0–10 | 4.75 | 2.3 |
| MOUG5 | Freshwater sediments | Peatland in France (Commana, France) | 0.2 | 5–15 | 6.7 | 8 |
| MOZ1 | Sediments from a pockmarck area | Mozambique Channel (Madagascar shore) | 762 | 2–4 | 8.0 | 8 |
| MOZ2 | Sediments from a pockmarck area | Mozambique Channel (Madagascar shore) | 762 | 4–6 | 8.0 | 8 |
| MOZ3 | Sediments from a pockmarck area | Mozambique Channel (Madagascar shore) | 762 | 6–11 | 8.0 | 8 |
| PAV60 | Anoxic water from a meromictic lake | Pavin lake (France) | 60 | – | 5.3 | 4.2 |
| PAV70 | Anoxic water from a meromictic lake | Pavin lake (France) | 70 | – | 5.3 | 4.75 |
| PAV80 | Anoxic water from a meromictic lake | Pavin lake (France) | 80 | – | 5.3 | 5 |
| PENF | Freshwater sediments | Tributary of the French river Penfeld (Brest, France) | 0.15 | 0–5 | 6.0 | 8 |
| XIA | Water from a geothermal hot spring | Hot spring in China (Xiamen Botanical Garden) | – | 0 | 6.3 | 82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cozannet, M.; Borrel, G.; Roussel, E.; Moalic, Y.; Allioux, M.; Sanvoisin, A.; Toffin, L.; Alain, K. New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments. Microorganisms 2021, 9, 30. https://doi.org/10.3390/microorganisms9010030
Cozannet M, Borrel G, Roussel E, Moalic Y, Allioux M, Sanvoisin A, Toffin L, Alain K. New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments. Microorganisms. 2021; 9(1):30. https://doi.org/10.3390/microorganisms9010030
Chicago/Turabian StyleCozannet, Marc, Guillaume Borrel, Erwan Roussel, Yann Moalic, Maxime Allioux, Amandine Sanvoisin, Laurent Toffin, and Karine Alain. 2021. "New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments" Microorganisms 9, no. 1: 30. https://doi.org/10.3390/microorganisms9010030
APA StyleCozannet, M., Borrel, G., Roussel, E., Moalic, Y., Allioux, M., Sanvoisin, A., Toffin, L., & Alain, K. (2021). New Insights into the Ecology and Physiology of Methanomassiliicoccales from Terrestrial and Aquatic Environments. Microorganisms, 9(1), 30. https://doi.org/10.3390/microorganisms9010030