3.1. Bacterial Community Analysis
In this study, we used amplicon sequencing to investigate the microbial distribution of endospore forming Firmicutes in two different sampling areas across Black Hills, SD, USA. Based on the identified 16S rRNA gene reads from the SDLC and SURF sediment metagenome, the total OTUs (Archaea and Bacteria) in SDLC and SURF were 66,738, and 106,144, respectively. This indicates that the total number of cells decreased as the soil temperature increased from 35–45 °C in the SURF sample to 55–65 °C in the SDLC sample. Thus, the difference in temperature could be one of the reasons for a significant difference in OTUs of both sites (p = 0.007).
In the SURF sample, the microbial communities were dominated by Bacteria with OTUs of 80,885 (Archaea was 25,259), which contributed approximately 73.2% of the total number of cells. However, in the 60 °C SDLC sample, Archaea became the dominant population with total OTUs of 48,367 (Bacteria was 18,371), and it accounted for more than 72.4% of the total number of cells. Moreover, between them, higher alpha diversity was observed in the SDLC (Shannon–Weaver index: Bacteria, 3.03; Archaea, 1.18), than in the SURF samples (Shannon–Weaver index: Bacteria, 1.91; Archaea, 1.06) (
Figure 1). This indicates that the higher temperature SDLC’s bacterial community profiles were substantially more complex than those of the lower temperature SURF site, with the archaeal diversity profoundly the same across both the sites. It is apparent in the literature that temperature, available organic matter, and C/N ratio are the key factors in the selection of microbial communities [
54]. In the SDLC compost, the levels of organic carbon, total carbon, and C/N ratios were higher than in the SURF samples (
Table 1). Further, in the self-heating compost compiles, there are a lot of organic matter, proteins, and other nutrients such as nitrogen and phosphorus, which support soil biological activity and functional diversity [
54]. Thus, the higher alpha diversity for the OTUs obtained for the metagenome of the SDLC compost could be due to the increase in complexity of the thermophilic communities with increases in levels of some physicochemical parameters like temperature, DO, water content, total organic matter, and total carbon in SDLC as compared to SURF.
Contrastingly, the unique deep subsurface of this mine is characterized by the presence of different water-soluble ions (sulfate, nitrate, iron) in soils [
49], with limited amounts of organic matter and total carbon. The SURF site is apparently the deepest mine (2.4 km deep) in the North America and had the largest gold deposit ever found in the Western Hemisphere [
55]. Today, the SURF site houses a world-renowned neutrino research lab, with geochemical characterization of soils revealing high amounts of toxic metals such as As, Cd, Co, Cr, Cu, Ni, Pb, Zn, and U [
55]. The presence of high metals, especially iron, indicates that the SURF sample where the organic energy sources are limited, can still support the growth of various chemotrophic microorganisms. However, the presence of low organic matter in the SURF site compared to the SDLC site can probably explain why the alpha diversity of bacterial communities in the in the SDLC samples was more enriched compared to the SURF samples.
Further, considering the fact that physiochemical properties in both the locations were very different from each other, we analyzed the samples for their β-Diversity. The Sorenson β-diversity dissimilarity index between the two samples at the phyla, class, and lower taxa was 0.101, 0.27, and 0.403, indicating the two communities were quite similar in sharing the common phyla, but dissimilar in terms of sharing the lower taxa. They also possessed significantly different community compositions. The community composition of phyla in both the SURF and SDLC samples is shown in
Table 2. In the whole community of SDLC, the three most abundant phyla were Firmicutes, Proteobacteria (γ-proteobacteria), and Actinobacteria, with relative abundances of approximately 86%, 6% and 6%, respectively. Within the whole SURF community, Firmicutes was still the dominant phylum at 69.5%, but the community was also significantly represented by Proteobacteria (α-proteobacteria and β-proteobacteria) and Bacteroidetes, which had a relative abundance of approximately 5%, and 2%, respectively. In addition, the SURF community had about 16% unclassified bacteria and 4% of its population with no hit.
Out of these, the members of Firmicutes, and Actinobacteria phyla are spore formers, and their combined higher total in SDLC (92%) than in SURF (71.5%) can be assigned to the higher prevailing temperature in SDLC compost (60 °C) over SURF sediments (45 °C). Firmicutes and Actinobacteria form resistant physiological stages that allow them to survive in hostile environments.
Nevertheless, despite so much variation in the two sampling sites in terms of their physiological properties, Firmicutes’ dominance in both the samples is clearly evident. Since the phylum Firmicutes members are endospore-forming aerobic or facultatively anaerobic bacteria, this property makes the phylum hardy in potentially harsh conditions and renders it a phenotypically and phylogenetically diverse taxon. Endospore formation allows these bacteria to be distributed into most of the habitats on Earth. While high-temperature conditions in the SDLC site can be attributed as a prime reason for the abundance of Firmicutes in the SDLC sample, the low carbon and moisture levels can explain their easy outgrowth against other phyla in the SURF subsurface environment.
Further, many of the members of Firmicutes are lignocellulolytic microorganisms as they can degrade tough lignocellulose in the plant biomass via the secretion of lignocellulases like cellulases, hemicellulases, and ligninolytic enzymes. Therefore, the abundance of Firmicutes in the SDLC sample rich in municipal solid wastes and yard waste is an anticipated event. Members of the Firmicutes phylum associated with biomass conversion are consistently abundant in both culture-based and 16S rRNA gene inventories of landfill sites, leading to the suggestion that they are the predominant degraders of biomass in landfill [
40]. However, the prevalence of Firmicutes in SURF samples with low organic matter content is a bit surprising. In the previous studies dealing with microbial community structures analysis in the subsurface environment, Proteobacteria, known to have sulfate reducers, nitrate reducers, and chemolithotrophic metabolizers, had been the dominant phyla in the majority of them [
55,
56]. However, the dominance of Firmicutes over Proteobacteria in this study, unaffected by sampling conditions having high metal concentrations, indicates a potential niche that remains unexplored for the members of Firmicutes. Gupta et al. 2018 demonstrated the ability of Firmicutes members to reduce sulfate and iron and presented a case for the role of Firmicutes for the in-situ bioremediation of an acid mine drainage impacted soil [
41]. Similar revelations have been made by authors of other studies [
57]. A large suit of metal resistance genes in the bacterial phyla Firmicutes was recently reported by Yong et al. (2018) [
58]. Therefore, ecologically, prevalence of Firmicutes in the Black soils of South Dakota signifies that this region represents a repository of biomass-degrading diversity that can be a rich source of new enzymes as well as metal resistance genes for environmental bioremediation. Further, these results support our hypothesis that members of Firmicutes are indeed versatile and can often be detected at a relatively high abundance in diverse environments, without strong dependence on soil composition or prevailing environmental conditions.
3.1.1. Community Composition of the Phylum Firmicutes
At the class level in SDLC, the most dominant class was Bacilli at 83%, followed by Clostridia at 3%. By contrast, the SURF sediments revealed a dominance of Clostridia at 46% over Bacilli at 20%. The shift in bacterial dominance from Bacilli to Clostridia in SURF sediments can be due to comparatively low oxygen conditions in this site, with Clostridia members preferring anaerobic conditions.
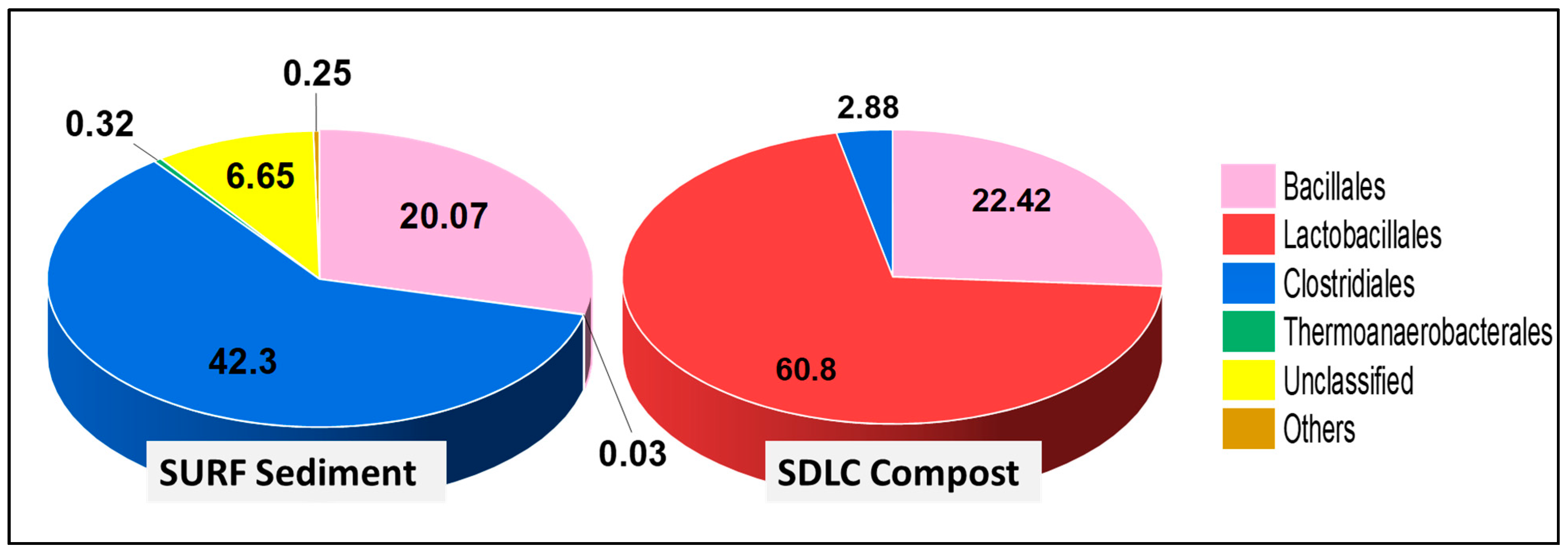
The dominance of members of Bacilli over Clostridia was evident down the phylogenetic levels in SDLC samples, where the order Lactobacillales (60.8%) was higher than Bacillales (22.4%) and Clostridiales (2.9%) (
Figure 2). Further, while the order Bacillales in SDLC was represented by a total of five families (Bacillaceae, Planococcaceae, Staphylococcaceae, Paenibacillaceae, and Thermoactinomycetaceae at 13%, 7.8%, 0.61%, 0.60%, and 0.29% respectively), Lactobacillales had a total representation from six families (
Aerococcaceae, Lactobacillaceae, Enterococcaceae, Carnobacteriaceae, Leuconostocaceae, and also Streptococcaceae at 21%, 16%, 13%, 6.1%, 4.5%, and 0.04%, respectively). By contrast, only two families represented Clostridiales (Clostridiaceae at 2.1%, and Eubacteriaceae at 0.78%). The prevalence of Lactobacillales over other orders in the SDLC sample can be correlated to the acidic pH conditions of the sample. Members of Lactobacillales are responsible for breaking down the organic waste materials into organic acids with an accompanied drop in pH in the compost [
59]. While the organic matter is being transformed, heat is released during the process, favoring actinomycetes and thermophilic bacteria such as
Bacillus sp. This explains the presence of Bacillales as the second most abundant order in the SDLC sample.
In comparison, within the SURF sample at the order level, there were no Lactobacillales at all, and Clostridiales (42%) still were prominent, with Bacillales at 20%. Here, there were only three prominent families within Bacillales represented by Bacillaceae, Planococcaceae, and Paenibacillaceae at 11%, 7.9% and 2.9%, respectively. However, the diversity of classes presented in SURF samples composing Clostridiales was relatively high, with a total of six families >0.1%, being Clostridiaceae (24%), Symbiobacteriaceae (7.6%), Ruminococcaceae (7.6%), Lachnospiraceae (1.2%), Peptococcaceae (0.34%), and Gracilibacteraceae (0.19%). This again highlights that while the diversity within the Bacillilales and Lactobacillales group was higher in the 60 °C SDLC samples, the 45 °C SURF samples had a higher diversity of Clostridiales and Thermoanaerobacterales, i.e., the anaerobic groups.
The leading bacterial communities for each sample was also analyzed at the genus level (
Figure 3). In the SDLC samples at the genus level, at least ten genera with >4% abundance were detected. Among them,
Aerococcus, Lactobacillus, Enterococcus, Desemzia, Solibacillus, Psychrobacillus, and
Weissella were the six most abundant genera with relative abundances of 21%, 15%, 13%, 5.9%, 4.9%, 4.7%, and 4.1%, respectively.
Bacillus, Geobacillus, Rummeliibacillus, Clostridium, and
Virgibacillus were the next five dominant genera with 3.6%, 2.5%, 2.5%, 2.1%, and 1.3%, respectively. In SDLC,
Lactobacillus, Paenibacillus, Leuconostoc, Gracilibacillus, Staphylococcus, Planococcus, Garciella, and
Planifilum were the minor genera detected with much lower average relative abundances (less than 1% across SDLC). By comparison, in the SURF samples, while the
Clostridium, Bacillus, Symbiobacterium, and
Solibacillus at 24%, 11%, 7.6%, and 7. 6%, were the three most abundant genera (more than 5%), the presence of
Paenibacillus (2.9%),
Acetivibrio (2.78%),
Ruminiclostridium (2.51%),
Ruminococcus (2.25%) were also detected in the sediments with more than 1% relative abundance.
In the SDLC site, the class Bacilli had a clear dominance and more diversity (11 genera with more than 1% OUTs) than in the SURF site, which had just two genera above 1% in the class Bacilli:
Paenibacillus, and
Solibacillus. For Clostridia in the SDLC sample (present at only 2.9%), only one genus,
Clostridium, was above 1%, but the diversity was higher in the SURF sample, where Clostridia (present at 46%) had five genera:
Clostridium, Acetovibrio, Ruminiclostridium, Ruminococcus, and
Symbiobacterium. Another feature of the SURF samples was the presence of unclassified family at 10% of the OTUs, and 3.5% of the OTUs being unclassclassified at the phylogenetic class level and lower levels. This comparison of the phylogenetic diversity at the level of genera between the samples highlights, again, the presence of higher bacterial diversity in the SDLC compost, as reflected in the large difference in the Shannon–Weaver index between the SDLC and SURF samples. Moreover, the abundance of Bacilli in the SDLC compost of this study, matches the results in some other studies, where it was mentioned that some members of the Bacillus genus have the ability to assimilate nitrogen and reduce the ammonium nitrogen loss during composting [
60].
3.1.2. Community Composition of the Phylum Actinobacteria
Beyond Firmicutes, the second phylum that is known to be spore producers and was present in the SDLC samples was Actinobacteria at 5.5% (
Figure 4). These actinobacteria belonged to a single class of Actinobacteria, subdivided into nine orders (>0.1% relative abundance), ten families (>0.1% relative abundance), and fourteen genera (>0.1% relative abundance). There was no presence of this phylum in the SURF sample, and this difference probably indicates the natural capacity of compost sites to enrich endospore-forming communities. Actinobacteria are bacteria with slow growth rates, but which have a greater capacity to degrade less biodegradable, complex organic compounds compared to other bacteria. They are often distributed in compost sites, whereby they can decompose complex mixtures of polymers in dead plants and animals, and make the carbon available to other prevailing communities [
59,
61].
3.1.3. Community Composition of Other Bacterial Phyla
Proteobacteria made up the second most dominant phylum in both the samples, with SDLC samples having 6.6% of Proteobacteria, and SURF sample having 5.4% of this phylum. Comparatively higher dominance of phylum Proteobacteria in the SURF site corroborated previous reports, as lineages of Proteobacteria are well-known to survive in low-nutrient environments, metal reduction, and metal resistance [
55]. Within this phylum, SDLC samples showed a dominance of γ-proteobacteria at 5.5% > β-proteobacteria (0.81%) > α-proteobacteria (0.33%). Within γ-proteobacteria, the dominant orders were represented by Pseudomonadales (3.4%) and Enterobacteriales (1.8%), and the three most represented families with more than 1% relative abundance were Moraxellaceae (2.0%), Enterobacteriaceae (1.8%), and Pseudomonadaceae (1.4%). At the genus level, the three most dominant genera having a representation of >1% were Acinetobacter (2.0%), Pseudomonas (1.4%), and Enterobacter (1.4%).
SURF samples showed a predominance of α-proteobacteria (4.1%) > β-proteobacteria (1.1%) > δ-proteobacteria (0.20%). All of these classes were solely represented by a single genus, order and family, i.e., α-proteobacteria by Rhodobacter (Order, Rhodobacterales; Family, Rhodobacteraceae) at 4.1%, β-proteobacteria by Alcaligenes (Order, Burkholderiales; Family, Alcaligenaceae) (1.14%), and γ-proteobacteria majorly by Pseudomonas (Order, Pseudomonadales; Family, Pseudomonadaceae) (0.2%). The analysis of the Shannon–Weaver index for species diversity of phylum Proteobacteria in SURF sediments of 0.64 vs. in that of SDLC of 2.21, again indicates the SDLC compost to be much more diverse in its representation across all the phyla.
Bacteroides was also present in both the samples, but while it was 0.56% in SDLC, the population was 1.9% in SURF. Beyond these phyla, SDLC samples just had Deinococcus-Thermus at 0.10%, with the rest of the population unclassified at 0.53%, and “No hits” at 0.61%. By comparison, SURF samples also had Cloroflexi (0.88%), Acidobacteria (0.65%), Spirochaetes (0.44%), Planctomycetes (0.37%), as some more of the representative phylum’s, with the unclassified phyla at 16.3%, and “No hits” being 4.3%. The presence of such a high a population of unclassified and no hits increases the probability of isolating novel species from the SURF site.
Since the primary aim of this work was to investigate the distribution of bacterial communities, in particular Firmicutes in the studied sites, we have not included in-depth discussion on distribution of arachael diversity in this paper. Howsoever, it may be noted that the archaea communities at both the sites exhibited lower diversity and were most closely affiliated to Euryarchaeota with 48.1%, and 57.6% of the reads in SDLC and SURF samples, respectively. The rest of the community was categorized as unclassified. Euryarchaeota is a diverse group which includes all members of the methanogens and certain thermophiles [
62]. While prevalence of methanogens in the SURF site has been reported previously by Rastogi et al. (2009) [
55], thermophilic methane oxidation has also been a reported activity within compost heaps [
63,
64]. Further investigation of the data and analysis of the distribution of archael thermophiles can be a rich source for additional investigations of thermophilic composting microbiology.
3.2. Identification of the Culturable Isolates
Since both the samples had Firmicutes as the dominant phyla, samples collected from both the sites were analyzed to evaluate the total thermophilic aerobic bacterial abundance. A total of 117 isolates with different colony morphologies were obtained from the enriched samples from the two sites (77 isolates from SDLC samples, numbered, LC1-77; and 40 isolates from SURF samples, SURF1-40). Coincidentally, all the isolated cells appeared Gram-positive, and based on their initial 16S rRNA gene analysis, it was found that there are a total of eight genera in the SDLC samples, with 43 species belonging to
Geobacillus, 20 species belonging to
Bacillus, four isolates of
Aneurinibacillus, three isolates belonging to
Ureibacillus, three isolates belonging to
Aeribacillus, two isolates within
Anoxybacillus, one isolate within
Parageobacillus, and one species that was recognized as
Thermoactinomyces. Out of these 77 isolates, 48 isolates were mere replicates of other representative species, and this bought the number of isolates from the SDLC samples that had different identities, as revealed by 16S identification, to 29 (17
Geobacillus, 6
Bacillus, two
Ureibacillus, one
Aeribacillus, one
Parageobacillus, one
Aneurinibacillus, one
Anoxybacillus, and one
Thermoactinomyces), representing 37.6% of the total isolates (
Supplementary Table S1).
On the other hand, in the SURF samples, isolates belonged to six different genera. Out of 40 isolates from SURF, 18 isolates were members of genus
Geobacillus, 12 isolates were from genus
Bacillus, four species belonged to
Paenibacillus, two species were from
Aeribacillus, one species was from
Ureibacillus, and three isolates were from genus
Parageobacillus. In these SURF samples, 26 isolates out of 40, representing 65% of the isolates, appeared different by 16S rRNA analysis (
Supplementary Table S2).
A more detailed analysis reveals total percentage of isolates from genus Geobacillus were higher in SDLC (58.61%) than SURF (30.70%); Bacillus were higher in SURF (34.60%) than SDLC (20.3%); Parageobacillus was higher in SURF (11.5%) than SDLC (3.4%); Aeribacillus were higher in SURF (7.7%), whereas Ureibacillus was equally represented within isolates from SURF and SDLC at 3.8% and 3.4% respectively. Further on, Paenibacillus was only present in SURF samples; while Aneurinibacillus, Anoxybacillus, and Thermoactinomyces species were only isolated from SDLC samples. Interestingly, after performing enrichment of compost and sediments and then performing isolations, members of Ureibacillus, Aeribacillus, Aneurinibacillus, Anoxybacillus, and Parageobacillus were also isolated. However, these genera were not detected in the metagenomic analysis.
3.2.1. Phylogenetic Relationship between Isolates from SDLC Sample Based on 16S rRNA Gene Sequence Comparison
Next, we compared the 16S rRNA gene sequences of 29 differential isolated strains from SDLC (
Figure 5) and 26 of the isolates from SURF (
Figure 6). We then checked the similarity index scores amongst their gene sequences using Clustal Omega. According to phylogenetic analysis of the 16S rRNA gene sequences of isolates from the SDLC samples, the isolated strains formed a total of fifteen groups, with ten groups (II, III, VII-XII, XIV and XV) having single species representation (
Figure 5). As regards the
Geobacillus species, based on isolates positioning on the phylogenetic tree, the
Geobacillus isolates were represented by Groups I-VI. Here, the group I had the isolates representing two different species
G. kaustophilus and
G. thermoleovorans. Within group I, there was also an unnamed species strains of
Geobacillus, numbered 3BC which had 97.61% similarity with
G. kaustophilus, and 96.87% similarity with
G. thermoleovorans. Group II and III had single representation with isolates that were identified as
G. zalihae, and
G. stearothermophilus, respectively. Group IV had the isolates belonging to clade
G. subterraneus with a % identity score of 99.16–99.80%
; group V isolates were identified as
G. thermodenitrificans with a % identity score of 98.45–99.12%; and group VI supposedly had the isolates found to be close to
G. toebii. In group VI, an isolate identified as
Parageobacillus toebii was found to create a sister grouping with
G. toebii, indicating that although
Geobacillus and
Parageobacillus are two ecologically diverse thermophilic genera within the phylum Firmicutes, their isolates are very close to each other phylogenetically and had a similarity score of 99.21%, thus putting a question mark on
Parageobacillus being classified as a separate genus. Also, group VI had two unnamed species strains of
Geobacillus, numbered E263, and TS3-9 with a similarity score of 97.20–99.86% between all the four members of the group, as revealed by multiple sequence alignment with Clustal Omega. Overall, the percentage similarity between all the seventeen SDLC isolates belonging to the genus
Geobacillus was 95.11–99.94%.
Since, from the SDLC samples, we also got the isolates identified to be belonging to different genus: SDLC-23S (as
Anoxybacillus kualawohkensis, group VII), SDLC-25S (as
Aeribacillus pallidus, group VIII), SDLC-4S (as
Aneurinibacillus migulanus, group IX), and SDLC-27S (as
Ureibacillus thermosphaericus, group X). We also checked the similarity score between these isolates belonging to different genera’s and found that percentage identity between the isolates was 85.65–93.77%. On the phylogenetic tree, while the genus
Anoxybacillus and
Aeribacillus were positioned more closely to members of
Geobacillus, the
Aneurinibacillus, and
Ureibacillus isolates were positioned more closely to members of
Bacillus on the phylogenetic tree (
Figure 5). Interestingly, despite being a separate genus, the
Anoxybacillus isolate SDLC-23S shared 94.10–96.89% identity with other isolates of
Geobacillus. The
Aeribacillus isolate SDLC-25S shared 93.31–95.82% identity with members of
Geobacillus in the SDLC sample. Likewise, SDLC-4S (
Aneurinibacillus sp.), and SDLC-27S (
Ureibacillus sp.) had 93.61–95.10%, and 94.80–96.98% identity with the other isolates of
Bacillus.Similar observations can be made within the isolates that were identified as Bacillus sp. by 16S rRNA analysis (placed in the groups XI-XIV). Herein, group XI had SDLC-15S identified to be B. kokeshiiformis, group XII had SDLC-33S identified to be B. smithii, SDLC 16S (B. pumilus) was present in group XIV, and three unnamed species of (Bacillus)—SDLC 7S, 11S and 13S came out to be forming a separate group (XIII). Overall, the percentage similarity between all the six SDLC isolates belonging to the genus Bacillus was 88.11–96.94%. Finally, the isolate representing Thermoactinomycetes vulgaris was more like an outlier within the isolates, with just 75% on the similarity score with rest of the isolates.
3.2.2. Phylogenetic Relationship between Isolates from SURF Sample Based on 16S rRNA Gene Sequence Comparison
While comparing the 16S rRNA gene sequences of 26 differential isolated strains from SURF, the strains were found to form twelve groups, with group VII-XI representing isolates that resembled Bacillus species, group I-IV represented isolates that resembled Geobacillus species, group V represented isolates that resembled Parageobacillus species, and group XII and VI had isolates resembling Paenibacillus, and Aeribacillus, respectively. As with SDLC samples, in terms of the similarity scores between the isolates’ 16S rRNA genes, the Geobacillus isolates from SURF also had the high percentage identity of 96.01–99.86% and, here also, the Parageobacillus strain was grouped well close to the Geobacillus group with 96.09–98.51% identity between it and rest of the Geobacillus isolates. Within the Geobacillus isolates, the isolates belonged to species thermodenitrificans, stearothermophilus, subterraneus, and thermoleovorans. While the SURF G. subterraneus member (SURF-3S) had >97% identity to members of G. thermodenitrificans, the isolate resembling G. stearothermophilus had 99% identity to G. thermoleovorans. Based on this analysis, we believe that Geobacillus isolates in SURF samples had representatives from clade G. thermodenitrificans, as well as G. thermoleovorans. This observation fits well with the fact that members of G. thermoleovorans are known more for fermentation in micro-aerobic conditions with no shaking and aeration, and the low oxygen conditions in the SURF site are probably responsible for their presence in this site rather than the SDLC site, which is comparatively aerobic.
Within the isolates representing Bacillus (groups VII-X), as in the case of Geobacillus, the percentage similarity between the isolates within each group was, on average, again was high at 99.92%, 97.13%, 97.52%, and 98.54%, respectively. Nevertheless, within the group XI, represented by Ureibacillus thermosphaericus and Bacillus kokeshiiformis, the percentage similarity was just 89.65 despite these two species being grouped. Moreover, the isolate resembling Bacillus kokeshiiformis had only 89.31–91.41% identity with the other Bacillus groups altogether. A similar observation was seen while analyzing the positioning of isolates resembling Bacillus smithii, which had 90.48–92.94% identity with the rest of the Bacillus members.
The SURF sample also had isolates belonging to Parageobacillus (group V), and Aeribacillus (group VI), with an intra-cluster identity of 90.28–92.87% and 95.23%, that formed groups between the Bacillus and Geobacillus groups. Both the groups Parageobacillus and Aeribacillus shared only 89.11–91.16% and 88.21–92.15% identity percentages with Bacillus and Geobacillus groups. This validates the classification of Parageobacillu, and Aeribacillus as a separate genus. On the other hand, three isolates belonging to genus Paenibacillus (SURF-11S, 7S, 16S) appeared far from Geobacillus, and this also qualifies Paenibacillus as a separate genus, descending away from Bacillus in the phylogenetic tree.
Overall, according to phylogenetic analysis of the 16S rRNA gene sequences from both SDLC and SURF isolates, while the level of intraspecific similarity between the sequences from Bacillus, Parageobacillus, Aeribacillus, and Paenibacillus groups was 89.65–99.54%, 89.11–91.16%, 88.21–92.15%, and 90.28–92.87%, respectively; the degree of interspecific similarity between the sequences from Geobacillus isolates was found to be high (95.11–100%); and this identity in the cases of sequences within the individual Geobacillus groups representing a particular species was extremely high (98.8–99.9%). These findings indicate that 16S rDNA sequences alone could not resolve the phylogenetic relationships between the Geobacillus species used. Although the low phylogenetic power at the species level and weak discriminatory power of 16S rRNA for Bacillus genera have been reported in other studies too; in this study, the high variation between the Bacillus members was only found if the species were identified as either B. smitthii, B. pumilus, or B. kokeshiiformi. The rest of the isolates belonging to genus Bacillus were still found to have a similarity score of 97.01–99.02% between them, which again did not permit reliable species differentiation.
Indeed, the bootstrap value at the nodes of a few clusters obtained was too low to induce much confidence. This again suggests that 16S rDNA sequences alone were not able to resolve the phylogenetic relationships between the Firmicutes isolated in this study, and it may be difficult in general to distinguish strains of the same genus with 16S rDNA information. The 16S rDNA sequence comparison might be effective for the analysis of genera and higher orders. The gyrB sequence comparison, on the other hand, may better for defining the phylogenetic relationships at the species level. Therefore, in this study, we also performed gyrB identification on the isolates, which has a more variable nucleotide sequence.
3.3. Identification of the Isolates Using gyrB rRNA Marker
We performed the identification analysis by gyrB with the same isolates, to check if more differentiation can be obtained for the strains. As done for 16S analysis, we first grouped the isolates based on results of gyrB, and we found that, where 16S analysis gave a total of 29 differentiated isolates from 77 isolates from LC samples, gyrB analysis gave 30 individual isolates (
Supplementary Table S3). However, with gyrB, more clarity was present in the groups formed within the phylogenetic tree.
3.3.1. Analysis of gyR by Phylogenetic Tree and Multiple Sequence Alignment for SDLC Isolates
Figure 7 below shows the phylogenetic tree that was obtained by aligning all the common isolates from the LC samples as identified by gyrB sequencing. Out of 77 isolates, 30 different strains have been identified, and these 30 isolates were found to form ten different groups (A–J) on a phylogenetic tree. Here, while group A consisted of all the
Geobacillus thermodenitrificans strains, and group C consisted of strains identified to be
Parageobacillus tobeii, including
Geobacillus sp. WCH70, Group B was an assortment of
Geobacillus strains identified to belong to many different species, i.e.,
G. subtterraneus, G. stereothermophilus, G. jurassicus, G. thermocatenulatus, G. zalihae, and
G. thermooleoverans. Here, an interesting grouping was seen for an isolate identified as
G. vulcani (group H) that was grouped with members of
Parageobacillus as a sister group. Overall, while within group A the similarity score stood between 97.23–99.95%, the similarity score within the isolates in group B was variable. While there was 99.92–99.85% identity between the three
Geobacillus subterraneus isolates, the identity score was 85.31–86.96% when the isolates belonged to a different
Geobacillus species. Furthermore, the similarity score between the isolates from
G. thermodenitrificans (group A) and isolates from group B (
G. subterraneus, G. tearothermophilus, G. jurassicus, G. thermocatenulatus, G. zalihae, and
G. thermoleovorans) was on an average between 90.59–85.83%. Consequently, it appears that gyrB is good enough to differentiate between different
Geobacillus species, whereas 16S rRNA failed to do so, as evident in the similarity scores being as high as 95.11–100%.
Furthermore, with gyrB sequencing, the genus Parageobacillus occurred separately from the clade Geobacillus, which just had an average 42–77% match with members of Geobacillus and qualified to be a separate genus. In fact, members of Parageobacillus occurred in two different groups in the phylogenetic tree (Group C and G) with identity between the groups of only 40.17% and represented by ideally different species, i.e., Parageobacillus toebii (in Group C) and Parageobacillus thermoglucosidans (in group G). Moreover, while group C had 76.92–77.19% identity across Geobacillus species represented in group A, group G shared only 41.68–43.17% identity with Geobacillus species represented in group B. Hence, with gyrB, strains previously identified as Geobacillus toebii and G. thermoglucosidans by 16S rRNA were identified as infact Parageobacillus toebii, and Parageobacillus thermoglucosidans. And gyrB again appears to be good enough to differentiate isolates of Geobacillus from those of Parageobacillus, where 16S rRNA again failed to do so, giving the identity between the two genera of 98.70–99.44%.
In the phylogenetic tree, identification of some of the Bacillus members as belonging to genus Kurthia and Lysinibacillus was another interesting feature obtained with gyrB sequencing. Within Group E, the isolates identified as Bacillus smithii, Kurthia populi, and Lysinibacillus sp. occurred together with a similarity score within the group of 69.48–77.38%. Intriguingly, the isolates of Kurthia and Lysinibacillus also occurred in two different groupings in the phylogenetic tree (groups E and I) with a similarity score of approximately 77.38–77.91% between Kurthia and Lysinibacillus in both the groups and 40–41.3% between the two groups. While group E isolates of Kurthia and Lysinibacillus existed as a sister group with the isolates identified to belong to genus Aeribacillus (group D); Group I isolate of Kurthia populli existed closer to Ureibacillus (Group J), with identity between groups of Aeribacillus and Ureibacillus of just 38–39%. Indeed, within Group J, the two Ureibacillus isolates, despite belonging to the same genus but different species, shared 86.71% similarity. Here again, gyrB proved to be capable of differentiating at the species level within the Ureibacillus, Bacillus, Kurthia, and Lysinibacillus genera.
3.3.2. Analysis of gyrB by Phylogenetic Tree and Multiple Sequence Alignment for SURF Isolates
Figure 8 below shows the phylogenetic tree that was obtained by aligning all the isolates (
Supplementary Table S4) from the SURF samples as identified by gyrB sequencing. Here, out of 44 isolates, a total of 27 different strains have been identified. These 27 isolates were found to form twelve different groups (A–L) on a phylogenetic tree, hence depicting more heterogeneity in the isolates obtained from the SURF samples than the SDLC samples and also in comparison to 16S profiling obtained for the same SURF isolates which just had nine groups. The isolates belonging to genus
Geobacillus were found to be divided into three groups H, I, and J, represented by characteristic species of
G. thermoleoverans,
G. subterraneus, and
G. thermodenitrificans. Within these three
Geobacillus groups, while the intraspecies percentage similarity of gyrase B identification within each group was 98.99–100%, the interspecies percentage identity amongst the
Geobacillus groups was 85.80–91.37%. The exception to this grouping was the
Geobacillus sp. strain WCH70 that, as in the earlier listing of gyrB from LC samples, was in the grouping with isolates of
Parageobacillus toebii in group K. Overall in the SURF samples, there was a total of 2 distinct types of
Parageobacillus, i.e.,
P. thermoglucosidasius (group K), and
P. toebii (group L), with percentage similarity between two distinct species, and hence in between K and L group of 42.24–43.26%, and intraspecies similarity of 89.11%, and 89.13% within group K, and within group M, respectively. A similar kind of index was present within the
Paenibacillus (group B), and
Aeribacillus (group E) isolates where the strains, despite belonging to the same genus but different species, shared 76.06%, 70.54–88.48%, and 96.11% similarity between them, respectively.
Unlike in SDLC samples where most of the Bacillus species were identified as belonging to genus Kurthia or Lysinibacillus, the Bacillus isolates in SURF samples more or less maintained their species as identified by 16S, except for Bacillus kokeshiiformis that was still recognized as belonging to Lysinibacter genus and it formed a separate grouping (group C) to that of other Bacillus isolates. The Bacillus isolates overall in the SURF isolates belonged to many different species like B. aerius, B. subtilis, B. velezensis, B. sonorensis, B. licheniformis, B. paralicheniformis, and B. smitthi, where the isolates belonging to different Bacillus species shared identity between them in the range 53.38–96.08%. This again indicates the versatility of gyrase B as a gene marker to differentiate between the species of Geobacillus, Parageobacillus, Paenibacillus, Aeribacillus, and Bacillus more precisely compared to 16S rRNA analysis.
One notable feature is that, while performing identification using gyrB, some of the isolates from SURF were showing results as multispecies DNA topoisomerase IV of Geobacillus and DNA topoisomerase IV of Ureibacillus, with no particular species. This may be due to gyrB being a less explored identification marker so far for the members of Firmicutes. In this work, the gyrB primers used were designed based on previous work by Tuotora et al. (2010) and was more specific for Firmicutes isolates.
Since the phylum Firmicutes members are endospore-forming aerobic or facultatively anaerobic bacteria, that makes its species hardy in potentially harsh conditions. This property makes this phylum a phenotypically and phylogenetically diverse taxon and allows these bacteria to be distributed into most of the habitats on Earth. However, due to the endospores themselves, the Firmicutes members are known to be resilient to many traditional methods of DNA isolation and thus potentially go undetected and underrepresented in metagenomic diversity surveys [
65,
66,
67]. Fillipidou et al., in their work in 2015, have highlighted this bias of metagenomic studies, with only a minor fraction of the community assigned to Firmicutes, while they evaluated the representation of endospore-forming Firmicutes in 73 published metagenomic datasets [
67]. Counter to this usual bias, we found Firmicutes to be the most dominant phylum of microbial life in both SDLC and SURF environments. Similarly, Actinobacteria (many of which have a spore stage) was detected as the second most dominant phylum in the SDLC sample, at 5.5%. We believe that the metagenomic analyses showing positive hits for the Firmicutes 16S rRNA gene in both of the two sites studied are due to the DNA extraction method we applied, and/or to the depth of sequencing that is hard to establish. The earliest and highly vital step in any metagenomic project is the extraction of DNA, which should be representative of all cells in the sample and involve a sufficient quality and amount for subsequent sequencing. The MINES method which makes use of a gentle preprocessing technique based on bead beating, combined with a multi-lytic polyzyme treatment, has been shown to be suitable enough for unbiased extraction of total genomic DNA from environmental samples for functional metagenomic studies [
43]. Hence, we consider that the “MINES” DNA isolation method used in this study [
43], along with the use of 16S rRNA metagenomic sequencing, facilitates the recovery of DNA representative of endospore-forming communities from the environmental samples, without any evident bias.
To further expand our understanding of the distribution of Firmicutes, a culture-dependent approach was applied to samples collected from the SDLC and SURF sites. This investigation resulted in the isolation of 77 isolates from SDLC samples, and 40 isolates from SURF samples, spanning five families (
Geobacillus,
Bacillus,
Aeribacillus, Paenibacillus, Parageobacillus) and also included a representative of Uncultured
Thermoactinomyces. Phylogenetic analysis of these isolates based on 16S rRNA gene sequence data indicated that isolates from both the samples were predominantly the strains belonging to genus
Geobacillus, representing 72.4% and 41.3% of the total isolates from the SDLC and SURF samples, respectively. It has been proven that
Geobacillus have a worldwide distribution in environments spanning hot springs to even cold climates, and this is almost certainly, in substantial portion, due to the adaptive features of their spores, which enable them to lie quiescent and achieve high population densities after accumulating gradually over an elongated timeline. The isolation of
Geobacillus representatives from the host compost (SDLC) at 60° and SURF sediments at 45 °C in high percentage, after a preconditioned enrichment, points to the excellent capability of this genus to colonize niches with varying temperatures, and low availability of nutritional compounds, due to their generally simple nutritional needs. The Black Hills region of South Dakota has never been studied earlier for the bacterial diversity of Firmicutes and its taxa. Hence, the isolation of strains of this phylum and genus
Geobacillus, found in high number in this study, should significantly add to the knowledge of the cultivation based microbial diversity of endospore formers. It is commonly understood that the culture-independent methods reveal more diversity of microbial communities in soil than culture-dependent methods. However, in this study, more
Geobacillus and Bacillus like bacterial genera were detected by the culture-dependent method rather than the culture independent metagenomic approach. In the first place, this discrepancy can be because of the specific enrichment procedure that was performed preceding the culture isolations for aerobic endospore forming Firmicutes members in this study. However, other researchers have also recently detected a higher percentage of cultivable
Bacillus-like genera from soils [
1,
68,
69]. Together, these findings indicate that DNA based metagenomic sequencing studies may often miss some information from some categories of Firmicutes genera that are present in the sites, but at low abundance. However, the explanation for these finding should be a matter of further research.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}