Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages



Abstract

1. Introduction
2. Materials and Methods
2.1. Culture Methods for THP-1 and RAW Cells
2.2. Culture Methods for Bacteria
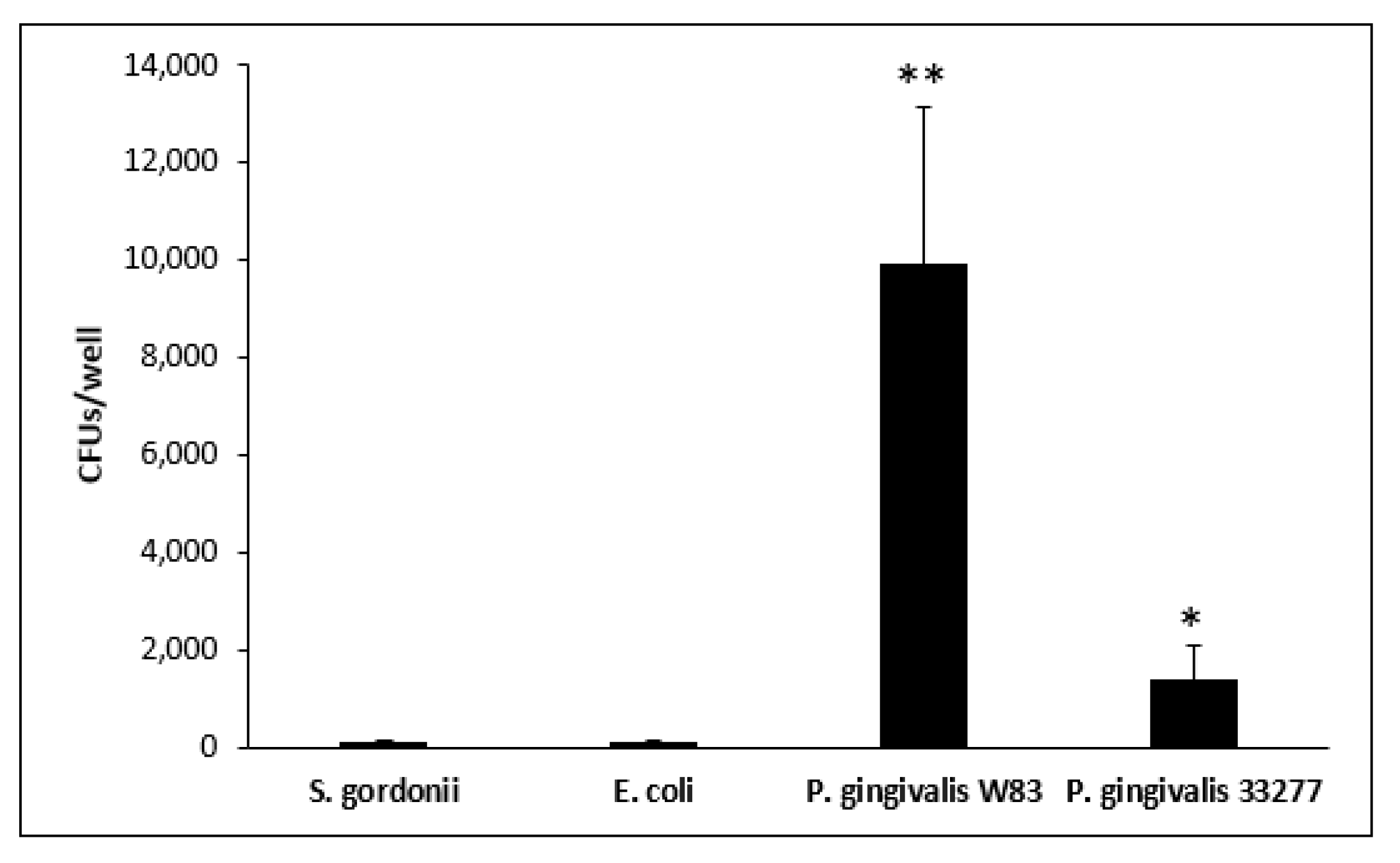
2.3. Measurements of Bacterial Internalization by CFU Counts
2.4. Measurements of Bacterial Escape by CFU Counts
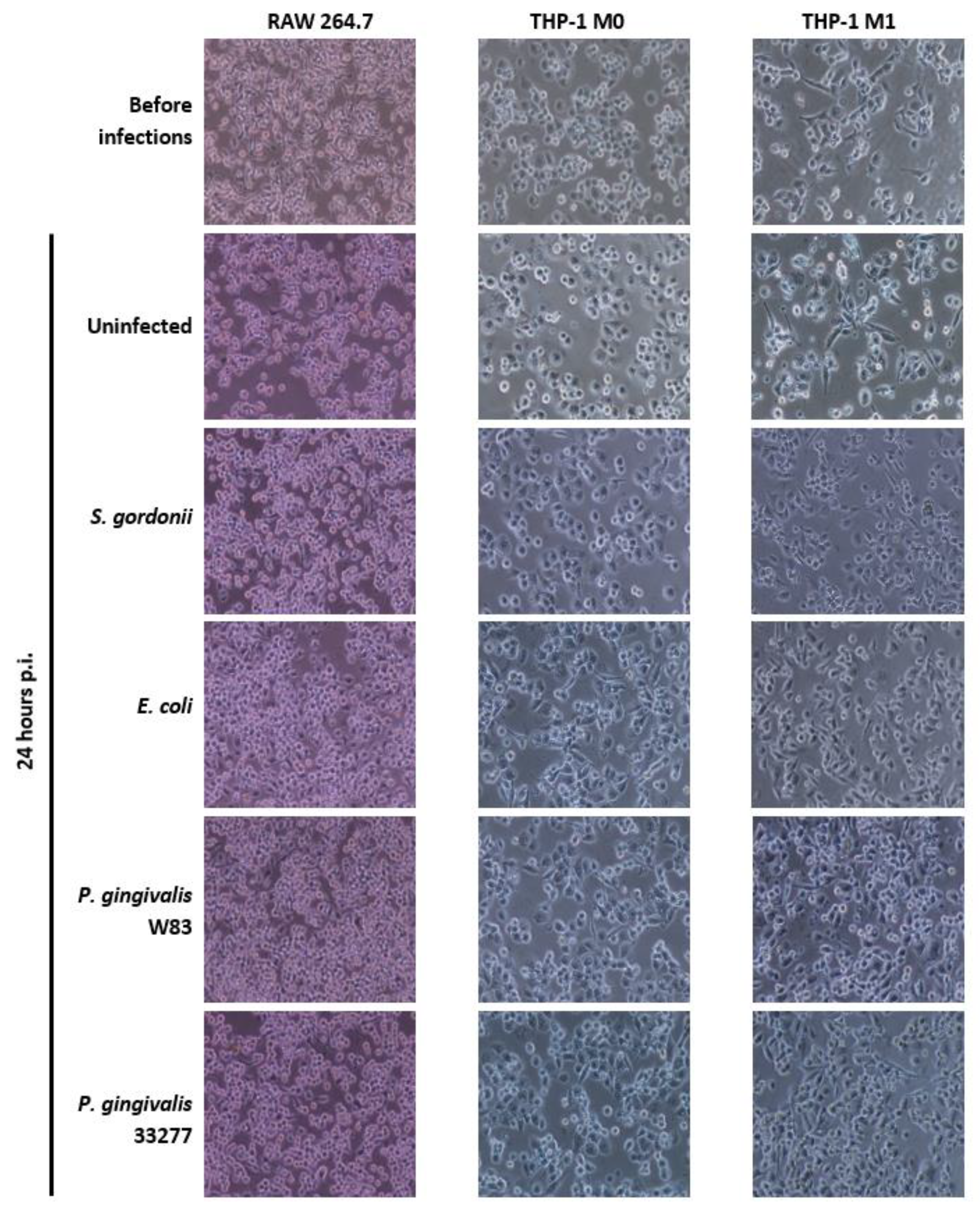
2.5. Assessment of Cellular Morphology
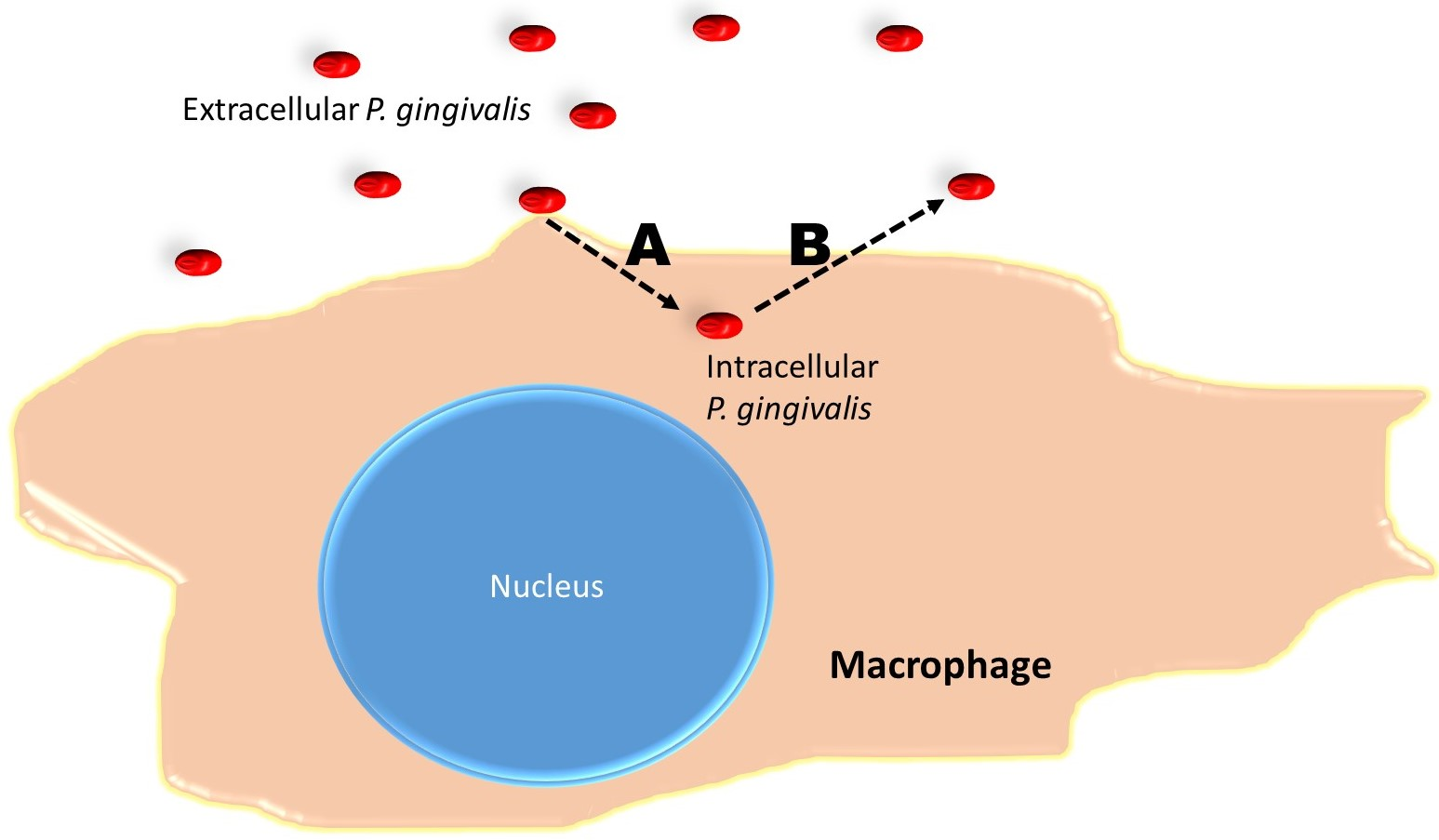
2.6. Measurements of Bacterial Escape and Re-Entry into Macrophages
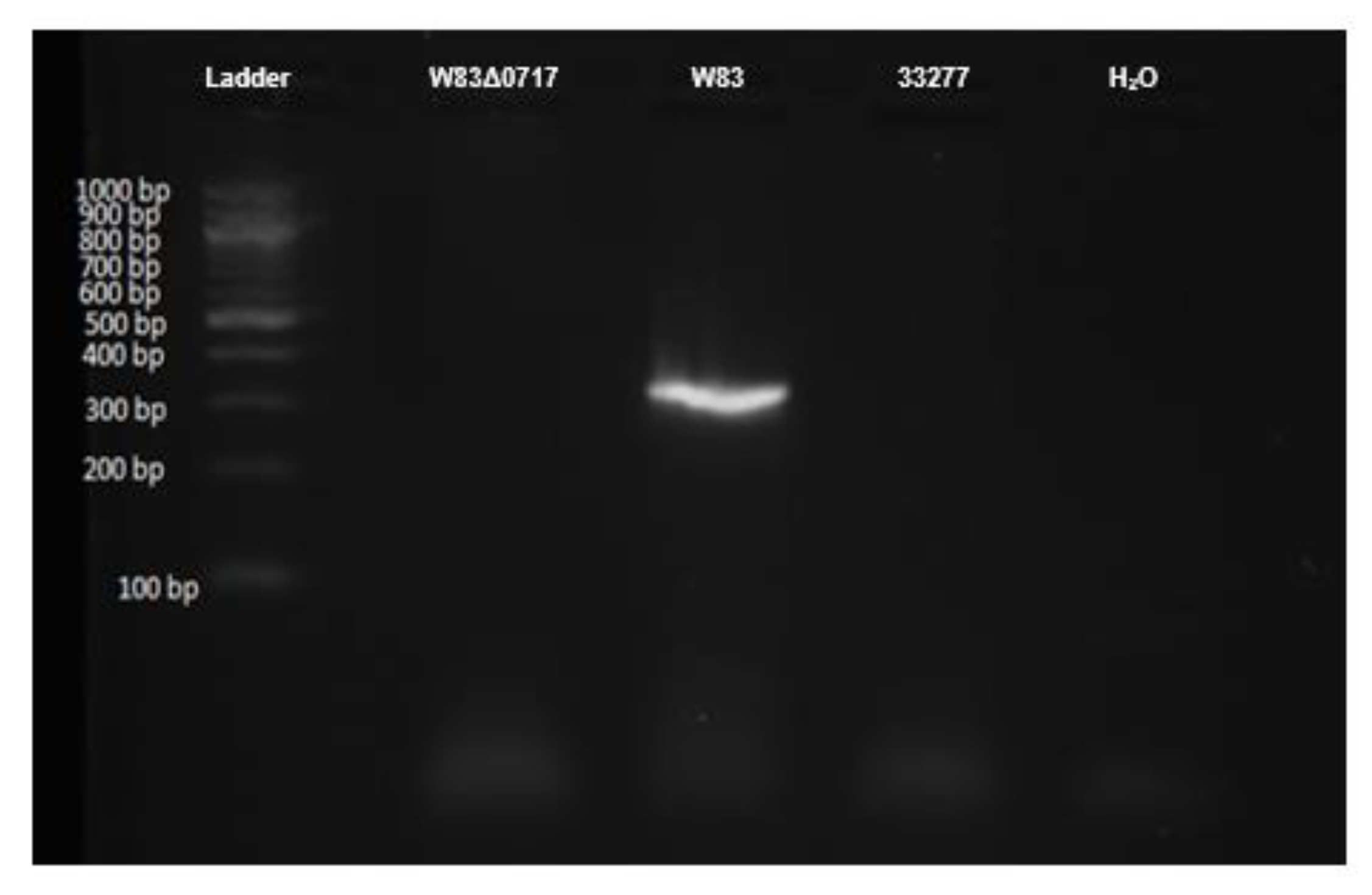
2.7. Genotyping of P. gingivalis W83Δ0717 Mutant Strain
2.8. P. gingivalis W83 and W83Δ0717 Escape from THP-1-Derived M1 Macrophages
2.9. Statistical Analysis
3. Results
3.1. Bacterial Growth in TSBY
3.2. Cellular Morphology of Mammalian Macrophages
3.3. Intracellular Bacteria
3.4. Escape Assay
3.5. Bacterial Escape and Re-Entry in M1 Cells
3.6. Confirmation of PG0717 Mutation
3.7. PG0717 Gene in Bacterial Escape
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eke, P.I.; Dye, B.A.; Wei, L.; Thornton-Evans, G.O.; Genco, R.J. CDC Periodontal Disease Surveillance workgroup: James Beck (University of North Carolina, Chapel Hill, USA), Gordon Douglass (Past President, American Academy of Periodontology), Roy Page (University of Washin Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 2012, 91, 914–920. [Google Scholar] [CrossRef]
- Periodontitis. MedlinePlus Medical Encyclopedia. Available online: https://medlineplus.gov/ency/article/001059.htm (accessed on 9 May 2020).
- Parahitiyawa, N.B.; Jin, L.J.; Leung, W.K.; Yam, W.C.; Samaranayake, L.P. Microbiology of odontogenic bacteremia: Beyond endocarditis. Clin. Microbiol. Rev. 2009, 22, 46–64. [Google Scholar] [CrossRef]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- The Immunological Genome Consortium; Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. The macrophage: Past, present and future. Eur. J. Immunol. 2007, 37, S9–S17. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Belfield, L.A.; Bennett, J.H.; Abate, W.; Jackson, S.K. Exposure to Porphyromonas gingivalis LPS during macrophage polarisation leads to diminished inflammatory cytokine production. Arch. Oral Biol. 2017, 81, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Tjiu, J.-W.; Chen, J.-S.; Shun, C.-T.; Lin, S.-J.; Liao, Y.-H.; Chu, C.-Y.; Tsai, T.-F.; Chiu, H.-C.; Dai, Y.-S.; Inoue, H.; et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Cochran, D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J. Periodontol. 2003, 74, 391–401. [Google Scholar] [CrossRef]
- Irwin, C.R.; Myrillas, T.T. The role of IL-6 in the pathogenesis of periodontal disease. Oral Dis. 1998, 4, 43–47. [Google Scholar] [CrossRef]
- Irwin, C.R.; Myrillas, T.T.; Traynor, P.; Leadbetter, N.; Cawston, T.E. The role of soluble interleukin (IL)-6 receptor in mediating the effects of IL-6 on matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1 expression by gingival fibroblasts. J. Periodontol. 2002, 73, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M. Professional and non-professional phagocytes: An introduction. Trends Cell Biol. 1995, 5, 85–87. [Google Scholar] [CrossRef]
- Kong, E.; Jabra-Rizk, M.A. The Great Escape: Pathogen Versus Host. PLoS Pathog. 2015, 11, e1004661. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res. Int. 2017, 2017, 1–18. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Levin, R.; Grinstein, S.; Canton, J. The life cycle of phagosomes: Formation, maturation, and resolution. Immunol. Rev. 2016, 273, 156–179. [Google Scholar] [CrossRef]
- Kinchen, J.M.; Ravichandran, K.S. Phagosome maturation: Going through the acid test. Nat. Rev. Mol. Cell Biol. 2008, 9, 781–795. [Google Scholar] [CrossRef]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; et al. A Potential New Pathway for Staphylococcus aureus Dissemination: The Silent Survival of S. aureus Phagocytosed by Human Monocyte-Derived Macrophages. PLoS ONE 2008, 3, e1409. [Google Scholar] [CrossRef]
- Stobernack, T.; du Teil Espina, M.; Mulder, L.M.; Palma Medina, L.M.; Piebenga, D.R.; Gabarrini, G.; Zhao, X.; Janssen, K.M.J.; Hulzebos, J.; Brouwer, E.; et al. A Secreted Bacterial Peptidylarginine Deiminase Can Neutralize Human Innate Immune Defenses. mBio 2018, 9, e01704-18. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immune Evasion Strategies of Porphyromonas gingivalis. J. Oral Biosci. 2011, 53, 233–240. [Google Scholar] [CrossRef]
- Dorn, B.R.; Dunn, W.A.; Progulske-Fox, A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 2001, 69, 5698–5708. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Roberts, J.S.; Choi, C.H.; Atanasova, K.R.; Yilmaz, Ö. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 2018, 9, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Tribble, G.D.; Lamont, R.J. Bacterial invasion of epithelial cells and spreading in periodontal tissue: Bacterial invasion of epithelial cells and spreading in periodontal tissue. Periodontology 2000 2010, 52, 68–83. [Google Scholar] [CrossRef]
- Brien-Simpson, N.M.; Veith, P.D.; Dashper, S.G.; Reynolds, E.C. Porphyromonas gingivalis gingipains: The molecular teeth of a microbial vampire. Curr. Protein Pept. Sci. 2003, 4, 409–426. [Google Scholar] [CrossRef]
- Miller, D.P.; Hutcherson, J.A.; Wang, Y.; Nowakowska, Z.M.; Potempa, J.; Yoder-Himes, D.R.; Scott, D.A.; Whiteley, M.; Lamont, R.J. Genes Contributing to Porphyromonas gingivalis Fitness in Abscess and Epithelial Cell Colonization Environments. Front. Cell. Infect. Microbiol. 2017, 7, 378. [Google Scholar] [CrossRef]
- Yoshino, T.; Laine, M.L.; van Winkelhoff, A.J.; Dahlén, G. Genotype variation and capsular serotypes of Porphyromonas gingivalis from chronic periodontitis and periodontal abscesses. FEMS Microbiol. Lett. 2007, 270, 75–81. [Google Scholar] [CrossRef]
- Karin Kristoffersen, A.; Solli, S.J.; Duy Nguyen, T.; Enersen, M. Association of the rgpB gingipain genotype to the major fimbriae (fimA) genotype in clinical isolates of the periodontal pathogen Porphyromonas gingivalis. J. Oral Microbiol. 2015, 7, 29124. [Google Scholar] [CrossRef]
- Rosan, B.; Lamont, R.J. Dental plaque formation. Microbes Infect. 2000, 2, 1599–1607. [Google Scholar] [CrossRef]
- Hajishengallis, G. The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 2014, 29, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, M.; Filardo, S.; Falasca, F.; Turriziani, O.; Sessa, R. Infectious Agents in Atherosclerotic Cardiovascular Diseases through Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 2459. [Google Scholar] [CrossRef] [PubMed]
- Shaik-Dasthagirisaheb, Y.B.; Mekasha, S.; He, X.; Gibson, F.C.; Ingalls, R.R. Signaling events in pathogen-induced macrophage foam cell formation. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef][Green Version]
- Wu, C.; Liu, C.; Luo, K.; Li, Y.; Jiang, J.; Yan, F. Changes in Expression of the Membrane Receptors CD14, MHC-II, SR-A, and TLR4 in Tissue-Specific Monocytes/Macrophages Following Porphyromonas gingivalis–LPS Stimulation. Inflammation 2018, 41, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, B.W.; Darveau, R.P. Porphyromonas gingivalis lipopolysaccharide: An unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol. Scand. 2001, 59, 131–138. [Google Scholar] [CrossRef]
- Hajishengallis, G.; McIntosh, M.L.; Nishiyama, S.-I.; Yoshimura, F. Mechanism and implications of CXCR4-mediated integrin activation by Porphyromonas gingivalis. Mol. Oral Microbiol. 2013, 28, 239–249. [Google Scholar] [CrossRef]
- McIntosh, M.L.; Hajishengallis, G. Inhibition of Porphyromonas gingivalis -induced periodontal bone loss by CXCR4 antagonist treatment: CXCR4 antagonist and bone loss. Microbiol. Oral Immunol. 2012, 27, 449–457. [Google Scholar] [CrossRef]
- Sun, J.; Nemoto, E.; Hong, G.; Sasaki, K. Modulation of stromal cell-derived factor 1 alpha (SDF-1α) and its receptor CXCR4 in Porphyromonas gingivalis-induced periodontal inflammation. BMC Oral Health 2017, 17, 26. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cha, G.S.; Kim, H.-J.; Kwon, E.-Y.; Lee, J.-Y.; Choi, J.; Joo, J.-Y. Porphyromonas gingivalis accelerates atherosclerosis through oxidation of high-density lipoprotein. J. Periodontal. Implant. Sci. 2018, 48, 60. [Google Scholar] [CrossRef]
- Fukasawa, A.; Kurita-Ochiai, T.; Hashizume, T.; Kobayashi, R.; Akimoto, Y.; Yamamoto, M. Porphyromonas gingivalis accelerates atherosclerosis in C57BL/6 mice fed a high-fat diet. Immunopharmacol. Immunotoxicol. 2012, 34, 470–476. [Google Scholar] [CrossRef]
- Hayashi, C.; Viereck, J.; Hua, N.; Phinikaridou, A.; Madrigal, A.G.; Gibson, F.C.; Hamilton, J.A.; Genco, C.A. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis 2011, 215, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Amano, A.; Inaba, H. Cardiovascular diseases and periodontal diseases. Clin. Calcium. 2012, 22, 43–48. [Google Scholar] [PubMed]
- Sharma, A.; Novak, E.K.; Sojar, H.T.; Swank, R.T.; Kuramitsu, H.K.; Genco, R.J. Porphyromonas gingivalis platelet aggregation activity: Outer membrane vesicles are potent activators of murine platelets. Oral Microbiol. Immunol. 2000, 15, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Garcia, R.; Heiss, G.; Vokonas, P.S.; Offenbacher, S. Periodontal disease and cardiovascular disease. J. Periodontol. 1996, 67, 1123–1137. [Google Scholar] [CrossRef]
- DeStefano, F.; Anda, R.F.; Kahn, H.S.; Williamson, D.F.; Russell, C.M. Dental disease and risk of coronary heart disease and mortality. BMJ 1993, 306, 688–691. [Google Scholar] [CrossRef]
- Kinane, D.F. Periodontal diseases’ contributions to cardiovascular disease: An overview of potential mechanisms. Ann. Periodontol. 1998, 3, 142–150. [Google Scholar] [CrossRef]
- Mealey, B.L. Influence of periodontal infections on systemic health. Periodontology 2000 1999, 21, 197–209. [Google Scholar] [CrossRef]
- Meyer, D.H.; Fives-Taylor, P.M. Oral pathogens: From dental plaque to cardiac disease. Curr. Opin. Microbiol. 1998, 1, 88–95. [Google Scholar] [CrossRef]
- Kumar, P.S. Oral microbiota and systemic disease. Anaerobe 2013, 24, 90–93. [Google Scholar] [CrossRef]
- Seymour, G.J.; Ford, P.J.; Cullinan, M.P.; Leishman, S.; Yamazaki, K. Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 2007, 13 (Suppl. 4), 3–10. [Google Scholar] [CrossRef]
- Zhou, X.; Han, J.; Liu, Z.; Song, Y.; Wang, Z.; Sun, Z. Effects of periodontal treatment on lung function and exacerbation frequency in patients with chronic obstructive pulmonary disease and chronic periodontitis: A 2-year pilot randomized controlled trial. J. Clin. Periodontol. 2014, 41, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Nakanishi, H. Lessons from Microglia Aging for the Link between Inflammatory Bone Disorders and Alzheimer’s Disease. J. Immunol. Res. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scannapieco, F.A. The oral microbiome: Its role in health and in oral and systemic infections. Clin. Microbiol. Newsl. 2013, 35, 163–169. [Google Scholar] [CrossRef]
- Borgnakke, W.S.; Ylöstalo, P.V.; Taylor, G.W.; Genco, R.J. Effect of periodontal disease on diabetes: Systematic review of epidemiologic observational evidence. J. Clin. Periodontol. 2013, 40, S135–S152. [Google Scholar] [CrossRef] [PubMed]
- Gaur, S.; Agnihotri, R. Alzheimer’s disease and chronic periodontitis: Is there an association?: Chronic periodontitis and Alzheimer’s disease. Geriatr. Gerontol. Int. 2015, 15, 391–404. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Harding, A.; Poole, S.; Kesavalu, L.; Crean, S. Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer’s Disease. Mediat. Inflamm. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Gibson, F.C., III. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front. Biosci. 2008, 13, 2041. [Google Scholar] [CrossRef]
- Isola, G.; Polizzi, A.; Santonocito, S.; Alibrandi, A.; Ferlito, S. Expression of Salivary and Serum Malondialdehyde and Lipid Profile of Patients with Periodontitis and Coronary Heart Disease. Int. J. Mol. Sci. 2019, 20, 6061. [Google Scholar] [CrossRef]
- Isola, G.; Alibrandi, A.; Currò, M.; Matarese, M.; Ricca, S.; Matarese, G.; Ientile, R.; Kocher, T. Evaluation of salivary and serum asymmetric dimethylarginine (ADMA) levels in patients with periodontal and cardiovascular disease as subclinical marker of cardiovascular risk. J. Periodontol. 2020, 91, 1076–1084. [Google Scholar] [CrossRef]
- Lin, L.; Pan, Y.; Li, C. Comparison between genes of highly toxic strain and minimally toxic strain of Porphyromonas gingivalis. Zhonghua Kou Qiang Yi Xue Za Zhi 2006, 41, 734–738. [Google Scholar]
- Reyes, L.; Eiler-McManis, E.; Rodrigues, P.H.; Chadda, A.S.; Wallet, S.M.; Bélanger, M.; Barrett, A.G.; Alvarez, S.; Akin, D.; Dunn, W.A.; et al. Deletion of Lipoprotein PG0717 in Porphyromonas gingivalis W83 Reduces Gingipain Activity and Alters Trafficking in and Response by Host Cells. PLoS ONE 2013, 8, e74230. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.H.; Reyes, L.; Chadda, A.S.; Bélanger, M.; Wallet, S.M.; Akin, D.; Dunn, W.; Progulske-Fox, A. Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: A comparative study. PLoS ONE 2012, 7, e52606. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.-H.; Huang, L.; Goodwin, J.S.; Dong, X.; Chen, C.-H.; Xie, H. Two Small Molecules Block Oral Epithelial Cell Invasion by Porphyromons gingivalis. PLoS ONE 2016, 11, e0149618. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Michel, R.; Cohen, J.; DeCarlo, A.; Kozarov, E. Intracellular survival and vascular cell-to-cell transmission of Porphyromonas gingivalis. BMC Microbiol. 2008, 8, 26. [Google Scholar] [CrossRef]
- Huang, Y.; Tian, C.; Li, Q.; Xu, Q. TET1 Knockdown Inhibits Porphyromonas gingivalis LPS/IFN-γ-Induced M1 Macrophage Polarization through the NF-κB Pathway in THP-1 Cells. Int. J. Mol. Sci. 2019, 20, 2023. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.K.; Pradhan, B.S.; Nayak, N.; Das, S.; Hsu, C.-D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Taciak, B.; Białasek, M.; Braniewska, A.; Sas, Z.; Sawicka, P.; Kiraga, Ł.; Rygiel, T.; Król, M. Evaluation of phenotypic and functional stability of RAW 264.7 cell line through serial passages. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Bélanger, M.; Rodrigues, P.; Progulske-Fox, A. Genetic manipulation of Porphyromonas gingivalis. Curr. Protoc. Microbiol. 2007, 5, 13C.2.1–13C.2.24. [Google Scholar] [CrossRef]
- Takeuchi, H.; Furuta, N.; Amano, A. Cell entry and exit by periodontal pathogen via recycling pathway. Commun. Integr. Biol. 2011, 4, 587–589. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, J.; Ren, G.; Zhang, Y.; Tan, X.; Yang, L. Punicalagin Prevents Inflammation in LPS-Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients 2019, 11, 2794. [Google Scholar] [CrossRef]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 577. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Mazhar, H.; Saleha, S.; Tipu, H.N.; Muhammad, N.; Abbas, M.N. Interferon-Gamma Improves Macrophages Function against M. tuberculosis in Multidrug-Resistant Tuberculosis Patients. Chemother. Res. Pract. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of Intracellular Shigella from Autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Xue, W.; Zhang, H.; Zhang, R.; Feldman, K.; Zhao, Q.; Zhang, S.; Shi, L.; Pavani, K.C.; Nian, W.; et al. MicroRNA-325-3p Facilitates Immune Escape of Mycobacterium tuberculosis through Targeting LNX1 via NEK6 Accumulation to Promote Anti-Apoptotic STAT3 Signaling. mBio 2020, 11. [Google Scholar] [CrossRef]
- Bacterial Defense against Phagocytosis. Available online: http://textbookofbacteriology.net/antiphago.html (accessed on 17 September 2020).
- Haraszthy, V.I.; Zambon, J.J.; Trevisan, M.; Zeid, M.; Genco, R.J. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 2000, 71, 1554–1560. [Google Scholar] [CrossRef]
- Azarpazhooh, A.; Leake, J.L. Systematic review of the association between respiratory diseases and oral health. J. Periodontol. 2006, 77, 1465–1482. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Ho, A.W. Potential associations between chronic respiratory disease and periodontal disease: Analysis of National Health and Nutrition Examination Survey III. J. Periodontol. 2001, 72, 50–56. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Papandonatos, G.D.; Dunford, R.G. Associations between oral conditions and respiratory disease in a national sample survey population. Ann. Periodontol. 1998, 3, 251–256. [Google Scholar] [CrossRef]
- Scannapieco, F.A.; Bush, R.B.; Paju, S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease. A systematic review. Ann. Periodontol. 2003, 8, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.I.; Nunn, M.E.; Vokonas, P.S. Epidemiologic associations between periodontal disease and chronic obstructive pulmonary disease. Ann. Periodontol. 2001, 6, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Boylan, R.J.; Kaslick, R.S.; Scannapieco, F.A.; Katz, R.V. Respiratory pathogen colonization of the dental plaque of institutionalized elders. Spec. Care Dent. 1999, 19, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.; Sparrow, D.; Cohen, M.; Vokonas, P.S.; Garcia, R.I. The association between alveolar bone loss and pulmonary function: The VA Dental Longitudinal Study. Ann. Periodontol. 1998, 3, 257–261. [Google Scholar] [CrossRef]
- Loos, B.G. Systemic Markers of Inflammation in Periodontitis. J. Periodontol. 2005, 76, 2106–2115. [Google Scholar] [CrossRef]
- Renvert, S.; Pettersson, T.; Ohlsson, O.; Persson, G.R. Bacterial Profile and Burden of Periodontal Infection in Subjects With a Diagnosis of Acute Coronary Syndrome. J. Periodontol. 2006, 77, 1110–1119. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I. The Epidemiology of Low-Grade Chronic Systemic Inflammation and Type 2 Diabetes. Diabetes Technol. Ther. 2006, 8, 7–17. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Offenbacher, S.; Wu, K.K.; Savage, P.J.; Heiss, G. Factor VIII and other hemostasis variables are related to incident diabetes in adults. The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care 1999, 22, 767–772. [Google Scholar] [CrossRef]
- Schmidt, M.I.; Duncan, B.B.; Sharrett, A.R.; Lindberg, G.; Savage, P.J.; Offenbacher, S.; Azambuja, M.I.; Tracy, R.P.; Heiss, G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): A cohort study. Lancet 1999, 353, 1649–1652. [Google Scholar] [CrossRef]
- Wang, X.; Bao, W.; Liu, J.; OuYang, Y.-Y.; Wang, D.; Rong, S.; Xiao, X.; Shan, Z.-L.; Zhang, Y.; Yao, P.; et al. Inflammatory Markers and Risk of Type 2 Diabetes: A systematic review and meta-analysis. Diabetes Care 2013, 36, 166–175. [Google Scholar] [CrossRef]
- King, G.L. The Role of Inflammatory Cytokines in Diabetes and Its Complications. J. Periodontol. 2008, 79, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s disease: Possible role of periodontal diseases. Alzheimer’s Dement. 2008, 4, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Watts, A.; Crimmins, E.M.; Gatz, M. Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2008, 4, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Petersen, P.E.; Kandelman, D.; Arpin, S.; Ogawa, H. Global oral health of older people—Call for public health action. Community Dent. Health 2010, 27, 257–267. [Google Scholar]
- Holmes, C.; Cotterell, D. Role of infection in the pathogenesis of Alzheimer’s disease: Implications for treatment. CNS Drugs 2009, 23, 993–1002. [Google Scholar] [CrossRef]
- Kamer, A.R.; Morse, D.E.; Holm-Pedersen, P.; Mortensen, E.L.; Avlund, K. Periodontal inflammation in relation to cognitive function in an older adult Danish population. J. Alzheimer’s Dis. 2012, 28, 613–624. [Google Scholar] [CrossRef]
- Batty, G.-D.; Li, Q.; Huxley, R.; Zoungas, S.; Taylor, B.-A.; Neal, B.; de Galan, B.; Woodward, M.; Harrap, S.-B.; Colagiuri, S.; et al. Oral disease in relation to future risk of dementia and cognitive decline: Prospective cohort study based on the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified-Release Controlled Evaluation (ADVANCE) trial. Eur. Psychiatry 2013, 28, 49–52. [Google Scholar] [CrossRef]
- Stein, P.S.; Desrosiers, M.; Donegan, S.J.; Yepes, J.F.; Kryscio, R.J. Tooth loss, dementia and neuropathology in the Nun study. J. Am. Dent. Assoc. 2007, 138, 1314–1322, quiz 1381–1382. [Google Scholar] [CrossRef]
- Kaye, E.K.; Valencia, A.; Baba, N.; Spiro, A.; Dietrich, T.; Garcia, R.I. Tooth loss and periodontal disease predict poor cognitive function in older men. J. Am. Geriatr. Soc. 2010, 58, 713–718. [Google Scholar] [CrossRef]
- Stewart, R.; Sabbah, W.; Tsakos, G.; D’Aiuto, F.; Watt, R.G. Oral health and cognitive function in the Third National Health and Nutrition Examination Survey (NHANES III). Psychosom. Med. 2008, 70, 936–941. [Google Scholar] [CrossRef]
- Isola, G.; Polizzi, A.; Iorio-Siciliano, V.; Alibrandi, A.; Ramaglia, L.; Leonardi, R. Effectiveness of a nutraceutical agent in the non-surgical periodontal therapy: A randomized, controlled clinical trial. Clin. Oral Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Kurita-Ochiai, T.; Kobayashi, R.; Suzuki, T.; Ando, T. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Kurita-Ochiai, T.; Yamamoto, M. Periodontal pathogens and atherosclerosis: Implications of inflammation and oxidative modification of LDL. Biomed. Res. Int. 2014, 2014, 595981. [Google Scholar] [CrossRef] [PubMed]
- Griffen, A.L.; Lyons, S.R.; Becker, M.R.; Moeschberger, M.L.; Leys, E.J. Porphyromonas gingivalis strain variability and periodontitis. J. Clin. Microbiol. 1999, 37, 4028–4033. [Google Scholar] [CrossRef]
- Papapanou, P.N.; Baelum, V.; Luan, W.M.; Madianos, P.N.; Chen, X.; Fejerskov, O.; Dahlén, G. Subgingival microbiota in adult Chinese: Prevalence and relation to periodontal disease progression. J. Periodontol. 1997, 68, 651–666. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| W83 | 33277 | References | |
|---|---|---|---|
| Major Fimbriae (genotype) | Yes (type IV in genes only) | Yes (type I) | [75] |
| Invasiveness | High | Low | [75] |
| Minor Fimbriae | No | Yes | [75,76,77] |
| PG0717 | Yes | No | [71] |
| Gingipains (genotype) | Kgp (I), RgpA (A), RgpB (NSSN, NYPN, NSSK—Possible association) | Kgp, RgpA no specific information on typing is available | [40,41] |
| Classification | Virulent | Avirulent | [78] |
| Capsule | Yes | No | [79] |
| Bacteria | Intracellular CFUs | Escaping CFUs a | Total CFU Escape b | Escaping and Re-Entering CFUs c |
|---|---|---|---|---|
| S. gordonii DL1 | 7030 | 125 (1.78%) 224 (3.18%) | 349 (4.96%) | 83 (23.78%) |
| E. coli OP50 | 10,167 | 696 (6.85%) 412 (4.05%) | 1108 (10.90%) | 94 (8.48%) |
| P. gingivalis W83 | 229,861 | 56,891 (24.75%) 97,530 (42.43%) | 154,421 (67.18%) | 9913 (6.42%) |
| P. gingivalis 33277 | 23,722 | 2000 (8.43%) 1912 (8.06%) | 3912 (16.49%) | 1358 (34.71%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werheim, E.R.; Senior, K.G.; Shaffer, C.A.; Cuadra, G.A. Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages. Microorganisms 2020, 8, 1432. https://doi.org/10.3390/microorganisms8091432
Werheim ER, Senior KG, Shaffer CA, Cuadra GA. Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages. Microorganisms. 2020; 8(9):1432. https://doi.org/10.3390/microorganisms8091432
Chicago/Turabian StyleWerheim, Erik R., Kevin G. Senior, Carly A. Shaffer, and Giancarlo A. Cuadra. 2020. "Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages" Microorganisms 8, no. 9: 1432. https://doi.org/10.3390/microorganisms8091432
APA StyleWerheim, E. R., Senior, K. G., Shaffer, C. A., & Cuadra, G. A. (2020). Oral Pathogen Porphyromonas gingivalis Can Escape Phagocytosis of Mammalian Macrophages. Microorganisms, 8(9), 1432. https://doi.org/10.3390/microorganisms8091432