Viral Abundance and Diversity of Production Fluids in Oil Reservoirs

 ,
,
Abstract
1. Introduction
2. Materials and Methods
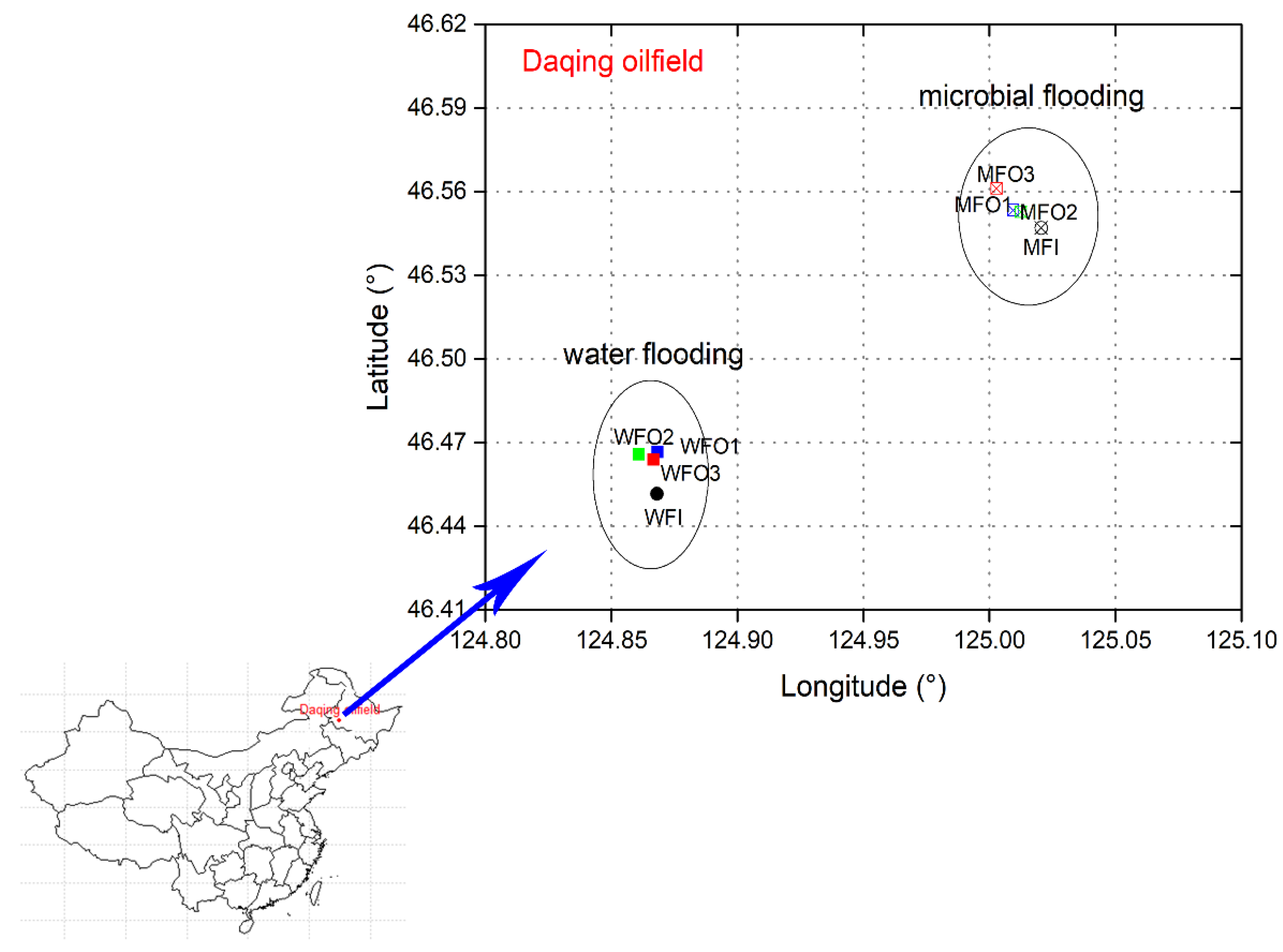
2.1. Site Description, Sample Collection and Pre-Treatment
2.2. Epifluorescence Microscopy
2.3. Virome Extraction and Sequencing
2.4. Virome Data Analysis
3. Results
3.1. VLP and Bacterial Abundance
3.2. Viruses to Bacteria Ratio
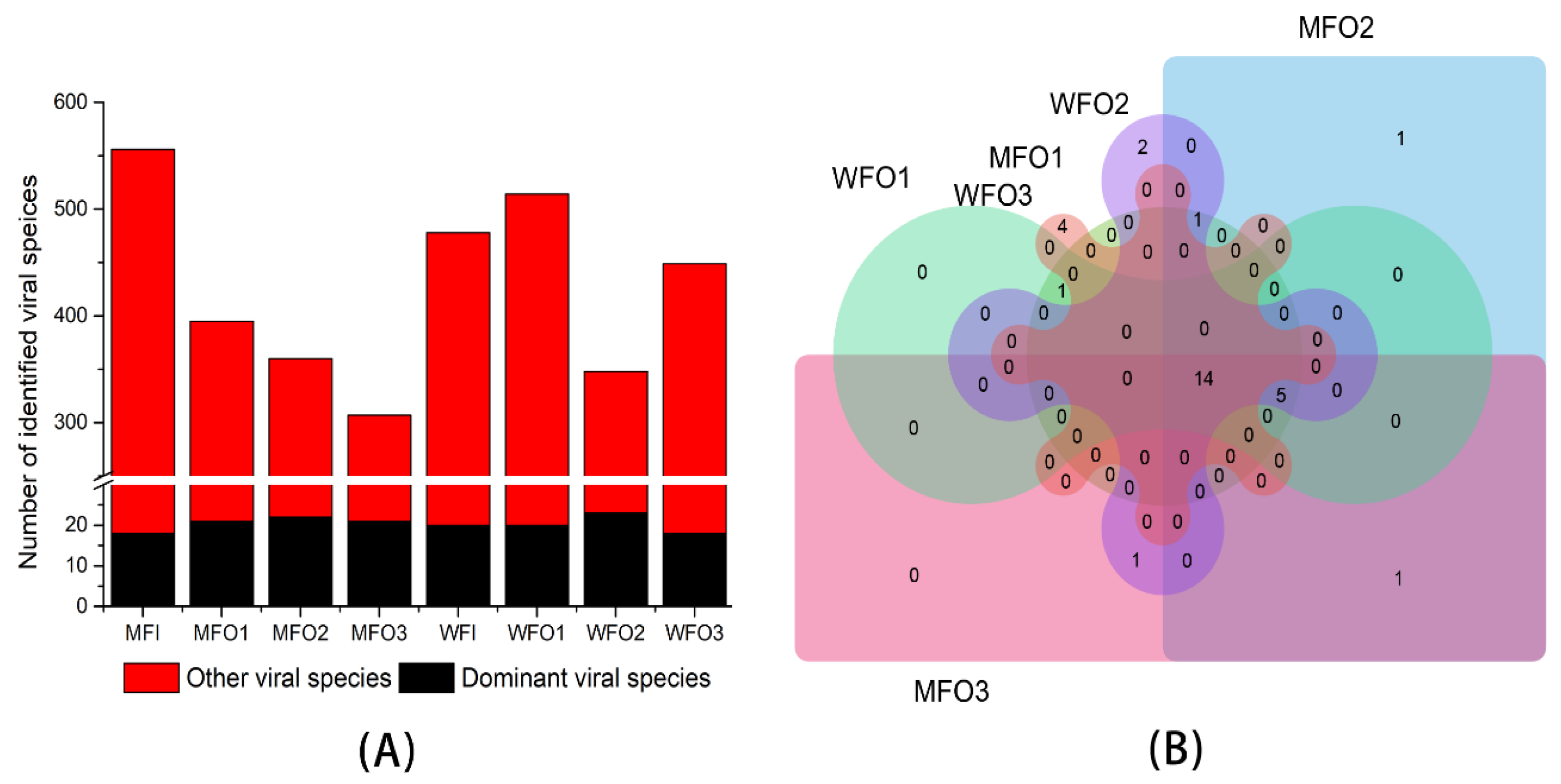
3.3. Viral Biodiversity
3.3.1. Viral Taxonomic Classification
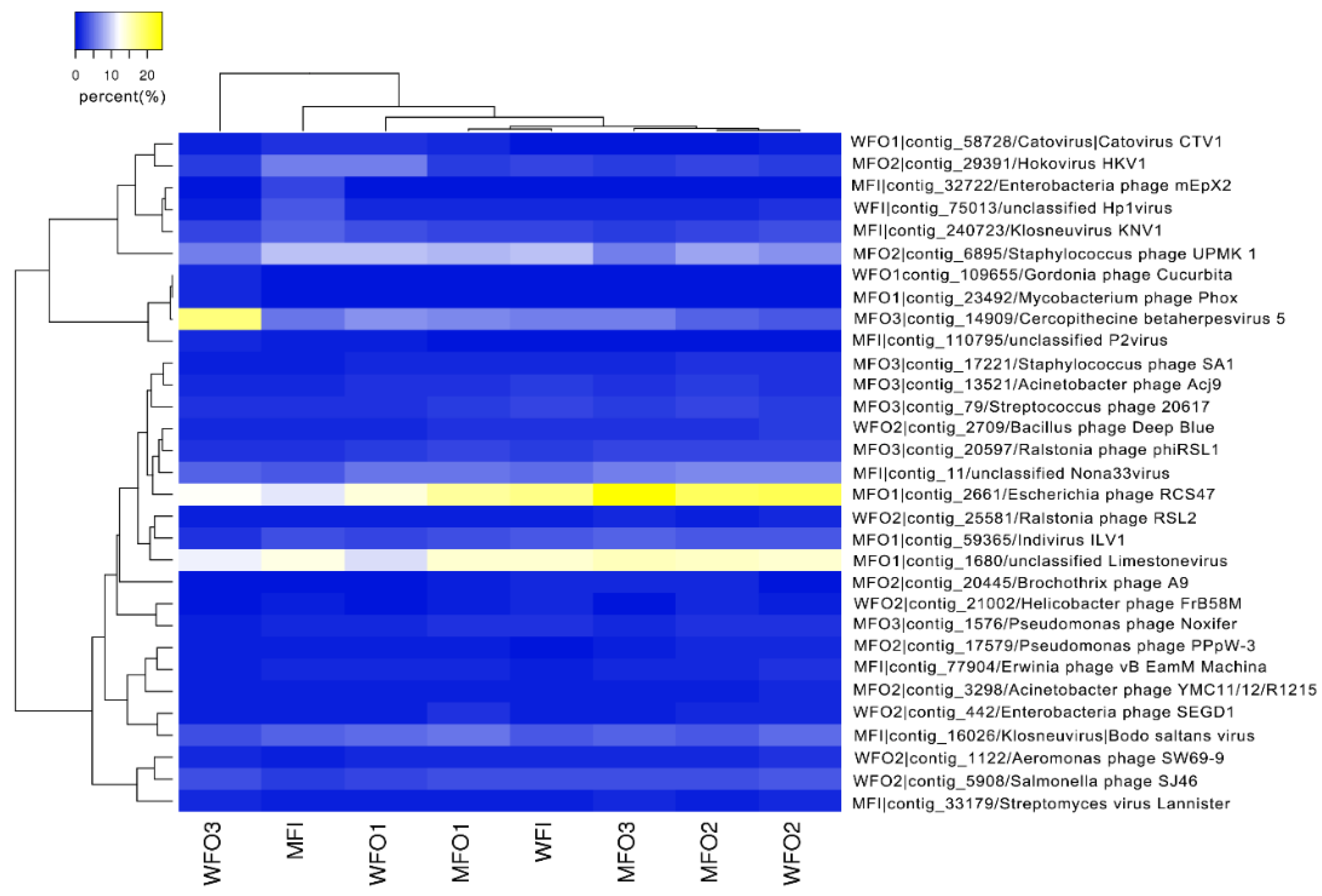
3.3.2. Viral Community Structure
3.3.3. Viral Diversity Analysis
4. Discussion
4.1. Viral and Bacterial Abundance in Oil Reservoir
4.2. Viral Community Diversity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ollivier, B.; Alazard, D. The Oil Reservoir Ecosystem. In Handbook of Hydrocarbon and Lipid Microbiology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 2259–2269. [Google Scholar]
- Liang, X.; Shi, R.; Radosevich, M.; Zhao, F.; Zhang, Y.; Han, S.; Zhang, Y. Anaerobic lipopeptide biosurfactant production by an engineered bacterial strain for in situ microbial enhanced oil recovery. RSC Adv. 2017, 7, 20667–20676. [Google Scholar] [CrossRef]
- Zhao, F.; Mandlaa, M.; Hao, J.; Liang, X.; Shi, R.; Han, S.; Zhang, Y. Optimization of culture medium for anaerobic production of rhamnolipid by recombinantPseudomonas stutzeri Rhl for microbial enhanced oil recovery. Lett. Appl. Microbiol. 2014, 59, 231–237. [Google Scholar] [CrossRef]
- Brussaard, C.P.D.; Wilhelm, S.W.; Thingstad, F.; Weinbauer, M.G.; Bratbak, G.; Heldal, M.; Kimmance, S.A.; Middelboe, M.; Nagasaki, K.; Paul, J.H.; et al. Global-scale processes with a nanoscale drive: The role of marine viruses. ISME J. 2008, 2, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wagner, R.E.; Zhuang, J.; Debruyn, J.M.; Wilhelm, S.W.; Liu, F.; Yang, L.; Staton, M.E.; Sherfy, A.C.; Radosevich, M. Viral abundance and diversity vary with depth in a southeastern United States agricultural ultisol. Soil Boil. Biochem. 2019, 137, 107546. [Google Scholar] [CrossRef]
- Liang, X.; Zhuang, J.; Löffler, F.E.; Zhang, Y.; Debruyn, J.M.; Wilhelm, S.W.; Schaeffer, S.M.; Radosevich, M. Viral and bacterial community responses to stimulated Fe(III)-bioreduction during simulated subsurface bioremediation. Environ. Microbiol. 2019, 21, 2043–2055. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and Nutrient Cycles in the Sea. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, Y.; Wommack, K.E.; Wilhelm, S.W.; Debruyn, J.M.; Sherfy, A.C.; Zhuang, J.; Radosevich, M. Lysogenic reproductive strategies of viral communities vary with soil depth and are correlated with bacterial diversity. Soil Boil. Biochem. 2020, 144, 107767. [Google Scholar] [CrossRef]
- Bi, L.; Yu, D.-T.; Du, S.; Zhang, L.-M.; Wu, C.-F.; Xiong, C.; Han, L.-L.; He, J.-Z. Diversity and potential biogeochemical impacts of viruses in bulk and rhizosphere soils. Environ. Microbiol. 2020. [Google Scholar] [CrossRef]
- Zeng, R.-Y.; Guo, X.; Zhang, R.; Qu, W.; Gao, B.; Zeng, R.-Y. Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome 2019, 7, 58. [Google Scholar] [CrossRef]
- Yu, D.-T.; Zheng, Y.; Zhang, L.; Han, L.-L. Viral metagenomics analysis and eight novel viral genomes identified from the Dushanzi mud volcanic soil in Xinjiang, China. J. Soils Sediments 2018, 19, 81–90. [Google Scholar] [CrossRef]
- Segobola, M.P.J.; Adriaenssens, E.M.; Tsekoa, T.; Rashamuse, K.; Cowan, D.A. Exploring Viral Diversity in a Unique South African Soil Habitat. Sci. Rep. 2018, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Han, L.-L.; Yu, D.-T.; Zhang, L.; Wang, J.-T.; He, J.-Z. Unique community structure of viruses in a glacier soil of the Tianshan Mountains, China. J. Soils Sediments 2016, 17, 852–860. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Lakshmi, M.; Kalpana, M.S.; Rao, P.L.S.; Patil, D.J.; Sudarshan, V.; Dayal, A.M. Geo-microbial and geochemical evidences in the near surface soils of Jamnagar sub-basin, Saurashtra, Gujarat, India: Implications to hydrocarbon resource potential. Geosci. J. 2012, 16, 455–467. [Google Scholar] [CrossRef]
- Wigington, C.H.; Sonderegger, D.; Brussaard, C.P.D.; Buchan, A.; Finke, J.F.; Fuhrman, J.A.; Lennon, J.T.; Middelboe, M.; Suttle, C.A.; Stock, C.; et al. Re-examination of the relationship between marine virus and microbial cell abundances. Nat. Microbiol. 2016, 1, 15024. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A Marine Viral Metagenomic Dataset and Associated Protein Clusters for Quantitative Viral Ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Roose-Amsaleg, C.; Fedala, Y.; Vénien-Bryan, C.; Garnier, J.; Boccara, A.-C.; Boccara, M. Utilization of interferometric light microscopy for the rapid analysis of virus abundance in a river. Res. Microbiol. 2017, 168, 413–418. [Google Scholar] [CrossRef]
- Martins, P.D.; Danczak, R.E.; Roux, S.; Frank, J.; Borton, M.A.; Wolfe, R.A.; Burris, M.N.; Wilkins, M.J. Viral and metabolic controls on high rates of microbial sulfur and carbon cycling in wetland ecosystems. Microbiome 2018, 6, 138. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Yu, Z.; Jin, J.; Liu, X.; Wang, G. Novel groups and unique distribution of phage phoH genes in paddy waters in northeast China. Sci. Rep. 2016, 6, 38428. [Google Scholar] [CrossRef]
- Voordouw, G.; Armstrong, S.M.; Reimer, M.F.; Fouts, B.; Telang, A.J.; Shen, Y.; Gevertz, D. Characterization of 16S rRNA genes from oil field microbial communities indicates the presence of a variety of sulfate-reducing, fermentative, and sulfide-oxidizing bacteria. Appl. Environ. Microbiol. 1996, 62, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Magot, M.; Ollivier, B.; Patel, B.K. Microbiology of Petroleum Reservoirs. Antonie Leeuwenhoek 2000, 77, 103–116. [Google Scholar] [CrossRef]
- Li, J.; Xue, S.; He, C.; Qi, H.; Chen, F.; Ma, Y. Effect of exogenous inoculants on enhancing oil recovery and indigenous bacterial community dynamics in long-term field pilot of low permeability reservoir. World J. Microbiol. Biotechnol. 2018, 34, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, W.; Wang, J.; Li, H.; Sun, J.; Ma, R.; Jiao, N.; Zhang, R. Tide driven microbial dynamics through virus-host interactions in the estuarine ecosystem. Water Res. 2019, 160, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Pfeiffer, J.K. Viruses and the Microbiota. Annu. Rev. Virol. 2014, 1, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.E.; Brazelton, W.J.; Baross, J. The Deep Viriosphere: Assessing the Viral Impact on Microbial Community Dynamics in the Deep Subsurface. Rev. Miner. Geochem. 2013, 75, 649–675. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Schwalbach, M. Viral Influence on Aquatic Bacterial Communities. Boil. Bull. 2003, 204, 192–195. [Google Scholar] [CrossRef]
- Kök, M.V. The Application of Water-flood Technology for Different Oil Fields. Energy Sources Part A Recover. Util. Environ. Eff. 2011, 33, 377–382. [Google Scholar] [CrossRef]
- Lazar, I.; Petrisor, I.G.; Yen, T.F. Microbial Enhanced Oil Recovery (MEOR). Pet. Sci. Technol. 2007, 25, 1353–1366. [Google Scholar] [CrossRef]
- She, H.; Kong, D.; Li, Y.; Hu, Z.; Guo, H. Recent Advance of Microbial Enhanced Oil Recovery (MEOR) in China. Geofluids 2019, 2019, 1–16. [Google Scholar] [CrossRef]
- Le, J.-J.; Wu, X.-L.; Wang, R.; Zhang, J.-Y.; Bai, L.-L.; Hou, Z.-W. Progress in pilot testing of microbial-enhanced oil recovery in the Daqing oilfield of north China. Int. Biodeterior. Biodegrad. 2015, 97, 188–194. [Google Scholar] [CrossRef]
- Hui, L.; Ai, M.; Han, S.; Guo, Y.; Li, N.; Shi, R.; Zeng, X.; Leng, S.; Zhao, J.; Liu, X.-B.; et al. Microbial diversity and functionally distinct groups in produced water from the Daqing Oilfield, China. Pet. Sci. 2012, 9, 469–484. [Google Scholar] [CrossRef]
- Wang, D.; Hou, Q.; Luo, Y.; Zhu, Y.; Fan, H. Feasibility Studies on CO2Foam Flooding EOR Technique after Polymer Flooding for Daqing Reservoirs. J. Dispers. Sci. Technol. 2014, 36, 453–461. [Google Scholar] [CrossRef]
- Thurber, R.L.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P. Analyzing fluorescence microscopy images with ImageJ. ImageJ 2014, 1, 195. [Google Scholar]
- John, S.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, K.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2018, 7, gix120. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.S.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Köstner, N.; Scharnreitner, L.; Jürgens, K.; Labrenz, M.; Herndl, G.J.; Winter, C. High viral abundance as a consequence of low viral decay in the Baltic Sea redoxcline. PLoS ONE 2017, 12, e0178467. [Google Scholar] [CrossRef] [PubMed]
- Payet, J.P.; McMinds, R.; Burkepile, D.E.; Thurber, R.L.V. Unprecedented evidence for high viral abundance and lytic activity in coral reef waters of the South Pacific Ocean. Front. Microbiol. 2014, 5, 493. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, M.; Liang, Y.; Zhang, Z.; Wang, F.; Jiang, X. Distribution of microbial populations and their relationship with environmental variables in the North Yellow Sea, China. J. Ocean Univ. China 2012, 11, 75–85. [Google Scholar] [CrossRef]
- Saxton, M.A.; Naqvi, N.S.; Rahman, F.; Thompson, C.P.; Chambers, R.M.; Kaste, J.M.; Williamson, K.E. Site-specific environmental factors control bacterial and viral diversity in stormwater retention ponds. Aquat. Microb. Ecol. 2016, 77, 23–36. [Google Scholar] [CrossRef]
- Lee, C.; Lim, J.; Heng, P. Viral Dynamics during a Phytoplankton Bloom Event. In Proceedings of the 14th Annual Meeting Geosciences Society (AOGS) 2017, Singapore, 6–11 August 2017. [Google Scholar]
- Huang, L.; Yu, L.; Luo, Z.; Song, S.; Bo, H.; Zheng, C. A Microbial-enhanced Oil Recovery Trial in Huabei Oilfield in China. Pet. Sci. Technol. 2014, 32, 584–592. [Google Scholar] [CrossRef]
- Rabus, R.; Fukui, M.; Wilkes, H.; Widdle, F. Degradative capacities and 16S rRNA-targeted whole-cell hybridization of sulfate-reducing bacteria in an anaerobic enrichment culture utilizing alkylbenzenes from crude oil. Appl. Environ. Microbiol. 1996, 62, 3605–3613. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Lakshmi, M.; Srinu, D.; Dayal, A.M. Bacteria as indicators for finding oil and gas reservoirs: A case study of the Bikaner-Nagaur Basin, Rajasthan, India. Pet. Sci. 2011, 8, 264–268. [Google Scholar] [CrossRef]
- Rasheed, M.A.; Lakshmi, M.; Rao, P.L.S.; Patil, D.J.; Dayal, A.M.; Sudarshan, V. Relevance of Pentane- and Hexane-Utilizing Bacterial Indicators for Finding Hydrocarbon Microseepage: A Study from Jamnagar Sub-basin, Saurashtra, Gujarat, India. Nat. Resour. Res. 2012, 21, 427–441. [Google Scholar] [CrossRef]
- Dellagnezze, B.; Vasconcellos, S.; Angelim, A.; Melo, V.M.M.; Santisi, S.; Cappello, S.; Oliveira, V. Bioaugmentation strategy employing a microbial consortium immobilized in chitosan beads for oil degradation in mesocosm scale. Mar. Pollut. Bull. 2016, 107, 107–117. [Google Scholar] [CrossRef]
- Wommack, K.E.; Colwell, R.R.; Saier, M.H. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Boil. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef]
- Parikka, K.J.; Le Romancer, M.; Wauters, N.; Jacquet, S. Deciphering the virus-to-prokaryote ratio (VPR): Insights into virus-host relationships in a variety of ecosystems. Boil. Rev. 2017, 92, 1081–1100. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Tay, Q.X.M.; Te, S.H.; Saeidi, N.; Goh, S.G.; Kushmaro, A.; Thompson, J.R.; Gin, K.Y.-H.; Xiaqiong, G. Geospatial distribution of viromes in tropical freshwater ecosystems. Water Res. 2018, 137, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Wang, D. Origins and challenges of viral dark matter. Virus Res. 2017, 239, 136–142. [Google Scholar] [CrossRef] [PubMed]
- De Cárcer, D.A.; Loópez-Bueno, A.; Pearce, D.A.; Alcami, A. Biodiversity and distribution of polar freshwater DNA viruses. Sci. Adv. 2015, 1, e1400127. [Google Scholar] [CrossRef] [PubMed]
- Sime-Ngando, T. Environmental bacteriophages: Viruses of microbes in aquatic ecosystems. Front. Microbiol. 2014, 5, 355. [Google Scholar] [CrossRef]
- Li, C.; Zhong, S.; Zhang, F.; Wang, Z.; Jiang, F.; Wan, Y. Response of microbial communities to supercritical CO2 and biogeochemical influences on microbially mediated CO2-saline-sandstone interactions. Chem. Geol. 2017, 473, 1–9. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bachani, P.; Jain, D.; Patidar, S.K.; Mishra, S. Extraction of potassium from K-feldspar through potassium solubilization in the halophilic Acinetobacter soli (MTCC 5918) isolated from the experimental salt farm. Int. J. Miner. Process. 2016, 152, 53–57. [Google Scholar] [CrossRef]
- Neu, U.; Mainou, B. Virus interactions with bacteria: Partners in the infectious dance. PLoS Pathog. 2020, 16, e1008234. [Google Scholar] [CrossRef]
- Sun, G.; Hu, J.; Wang, Z.; Li, X.; Wang, W. Dynamic investigation of microbial activity in microbial enhanced oil recovery (MEOR). Pet. Sci. Technol. 2018, 36, 1265–1271. [Google Scholar] [CrossRef]
- Øvreås, L.; Bourne, D.; Sandaa, R.; Casamayor, E.O.; Benlloch, S.; Goddard, V.; Smerdon, G.; Heldal, M.; Thingstad, T. Response of bacterial and viral communities to nutrient manipulations in seawater mesocosms. Aquat. Microb. Ecol. 2003, 31, 109–121. [Google Scholar] [CrossRef]
- Winter, C.; Matthews, B.; Suttle, C.A. Effects of environmental variation and spatial distance on Bacteria, Archaea and viruses in sub-polar and arctic waters. ISME J. 2013, 7, 1507–1518. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Number of Reads (PE) | Total Number of Bases (Mb) | Total Number of Contig | GC Content (%) | Longest Contig Size (bp) | Shortest Contig Size (bp) | % Total Abundance | |
|---|---|---|---|---|---|---|---|---|
| Annotated as Virus | Unclassified | |||||||
| WFI | 34089873 | 311.56 | 253712 | 41.68 | 232481 | 300 | 36.64 | 63.36 |
| WFO1 | 30623735 | 116.67 | 98943 | 47.25 | 567560 | 300 | 27.72 | 72.28 |
| WFO2 | 24602901 | 46.28 | 44000 | 39.73 | 96276 | 300 | 56.37 | 43.63 |
| WFO3 | 24137426 | 32.55 | 26033 | 45.87 | 234881 | 300 | 21.93 | 78.07 |
| MFI | 28158702 | 156.98 | 132111 | 40.45 | 290374 | 300 | 26.36 | 73.634 |
| MFO1 | 42093578 | 198.6 | 166617 | 45.31 | 316529 | 300 | 31.56 | 68.44 |
| MFO2 | 26647904 | 41.41 | 41931 | 41.35 | 62494 | 300 | 60.90 | 39.10 |
| MFO3 | 45944830 | 203.86 | 170199 | 48.38 | 425897 | 300 | 75.37 | 24.63 |
| Sample | Percentage of Annotated Contigs (%) | Viral Percentage in Annotated Contigs (%) | Non-virus Percentage in Annotated Contigs (%) | Phage Percentage in Annotated Viral Contigs (%) |
|---|---|---|---|---|
| WFI | 50.78 | 36.64 | 63.36 | 21.64 |
| WFO1 | 37.3 | 27.72 | 72.28 | 23.33 |
| WFO2 | 72.01 | 56.37 | 43.63 | 26.45 |
| WFO3 | 27.78 | 21.93 | 78.07 | 27.46 |
| MFI | 38.05 | 26.36 | 73.64 | 19.24 |
| MFO1 | 38.36 | 31.56 | 68.44 | 29.91 |
| MFO2 | 77.98 | 60.90 | 39.1 | 26.79 |
| MFO3 | 92.27 | 75.37 | 24.63 | 29.00 |
| Virus | Host |
|---|---|
| Escherichia phage RCS47 | Escherichia coli |
| Cercopithecine betaherpesvirus 5 | Cercopithecus |
| Salmonella phage SJ46 | Salmonella enterica subsp. enterica serovar Indiana |
| Ralstonia phage phiRSL1 | Ralstonia solanacearum |
| Streptococcus phage 20617 | Streptococcus thermophilus DSM 20617 |
| Acinetobacter phage Acj9 | Acinetobacter johnsonii |
| Pseudomonas phage Noxifer | Pseudomonas fluorescens SBW25 |
| Bacillus phage Deep Blue | Bacillus cereus BtB2-4 |
| Staphylococcus phage SA1 | Staphylococcus aureus |
| Erwinia phage vB EamM Machina | Erwinia amylovora |
| Brochothrix phage A9 | Brochothrix thermosphacta |
| Ralstonia phage RSL2 | Ralstonia |
| Pseudomonas phage PPpW-3 | Pseudomonas plecoglossicida |
| Gordonia phage Cucurbita | Gordonia terrae |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, L.; Liang, X.; Shi, R.; Li, P.; Zhao, J.; Li, G.; Wang, S.; Han, S.; Radosevich, M.; Zhang, Y. Viral Abundance and Diversity of Production Fluids in Oil Reservoirs. Microorganisms 2020, 8, 1429. https://doi.org/10.3390/microorganisms8091429
Zheng L, Liang X, Shi R, Li P, Zhao J, Li G, Wang S, Han S, Radosevich M, Zhang Y. Viral Abundance and Diversity of Production Fluids in Oil Reservoirs. Microorganisms. 2020; 8(9):1429. https://doi.org/10.3390/microorganisms8091429
Chicago/Turabian StyleZheng, Liangcan, Xiaolong Liang, Rongjiu Shi, Ping Li, Jinyi Zhao, Guoqiao Li, Shuang Wang, Siqin Han, Mark Radosevich, and Ying Zhang. 2020. "Viral Abundance and Diversity of Production Fluids in Oil Reservoirs" Microorganisms 8, no. 9: 1429. https://doi.org/10.3390/microorganisms8091429
APA StyleZheng, L., Liang, X., Shi, R., Li, P., Zhao, J., Li, G., Wang, S., Han, S., Radosevich, M., & Zhang, Y. (2020). Viral Abundance and Diversity of Production Fluids in Oil Reservoirs. Microorganisms, 8(9), 1429. https://doi.org/10.3390/microorganisms8091429