Metagenomic and Metatranscriptomic Study of Microbial Metal Resistance in an Acidic Pit Lake

 , , and
, , and
Abstract
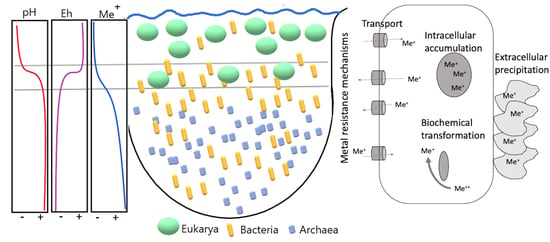
1. Introduction
2. Materials and Methods
2.1. Geologic Setting
2.2. Sample Collection, Physico-Chemical Profiling/Field Data Acquisition and DNA/RNA Extraction
2.3. Metagenomics and Metatranscriptomics Sequencing
2.4. Whole Community Metagenome and Metatranscriptome Processing
2.5. Metagenome-Assembled Genome Processing
2.6. Metal Resistance Gene Database
2.7. Data Availability
3. Results and Discussion
3.1. Toxic Potency Factors
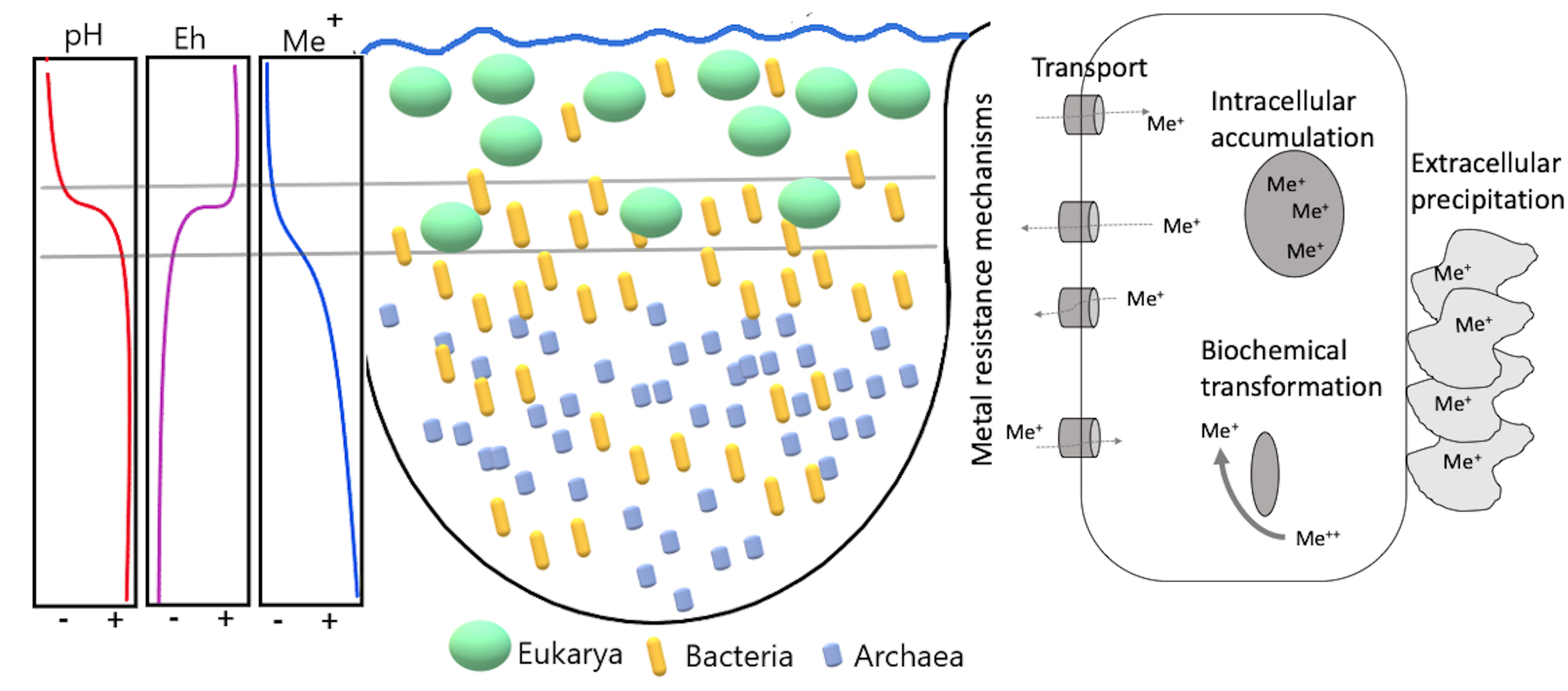
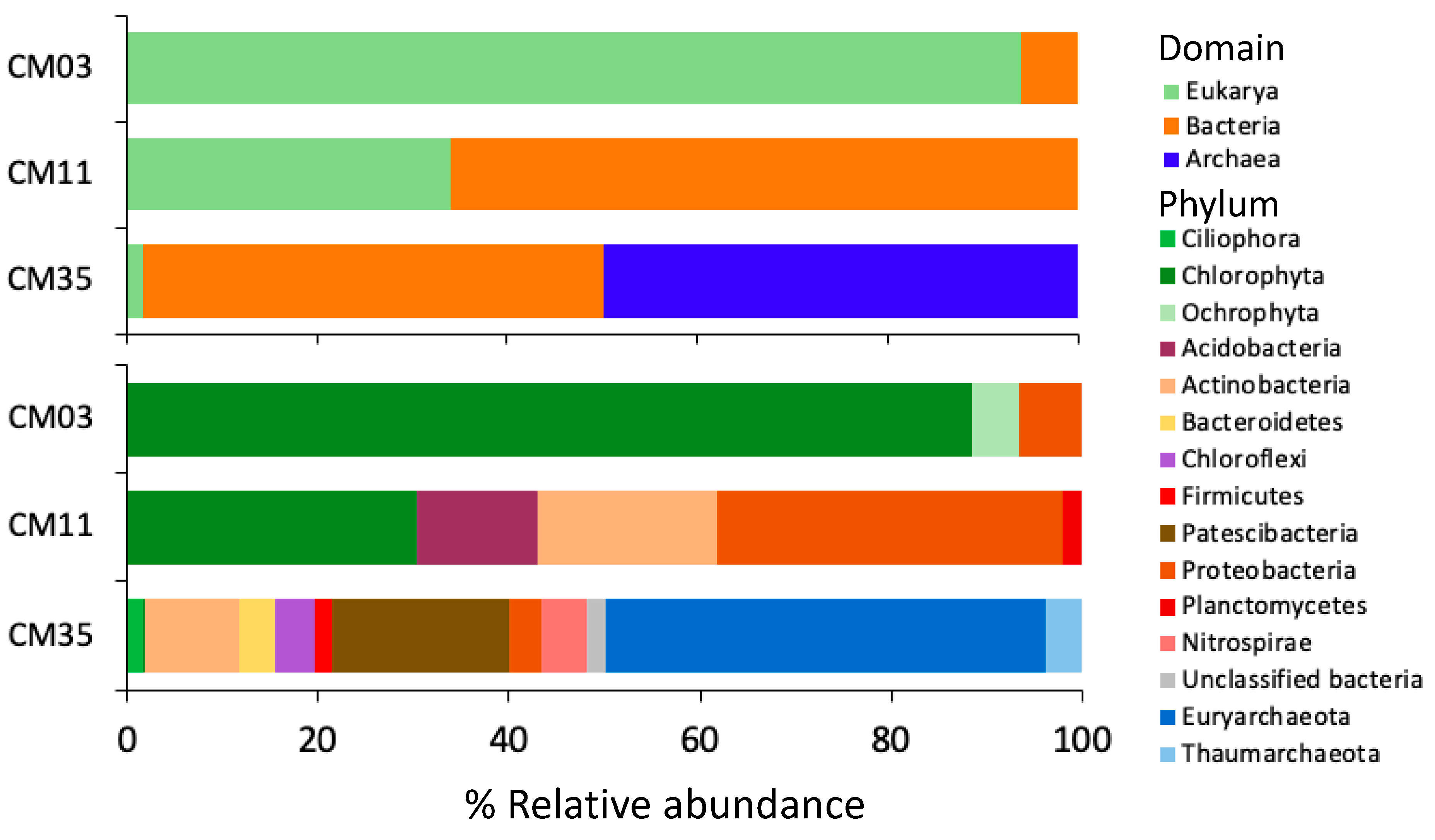
3.2. Microbial Diversity
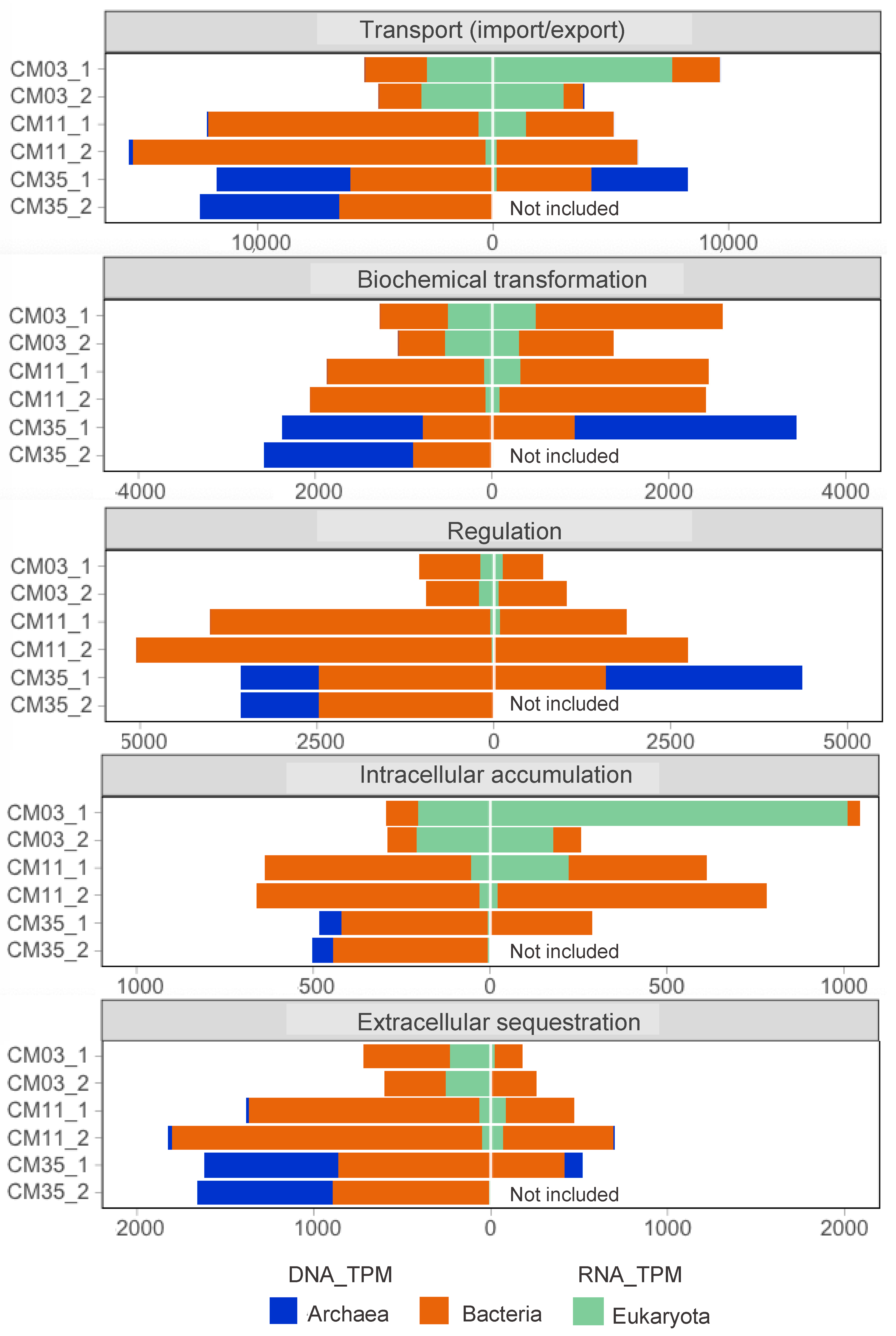
3.3. Metal Resistance Mechanisms
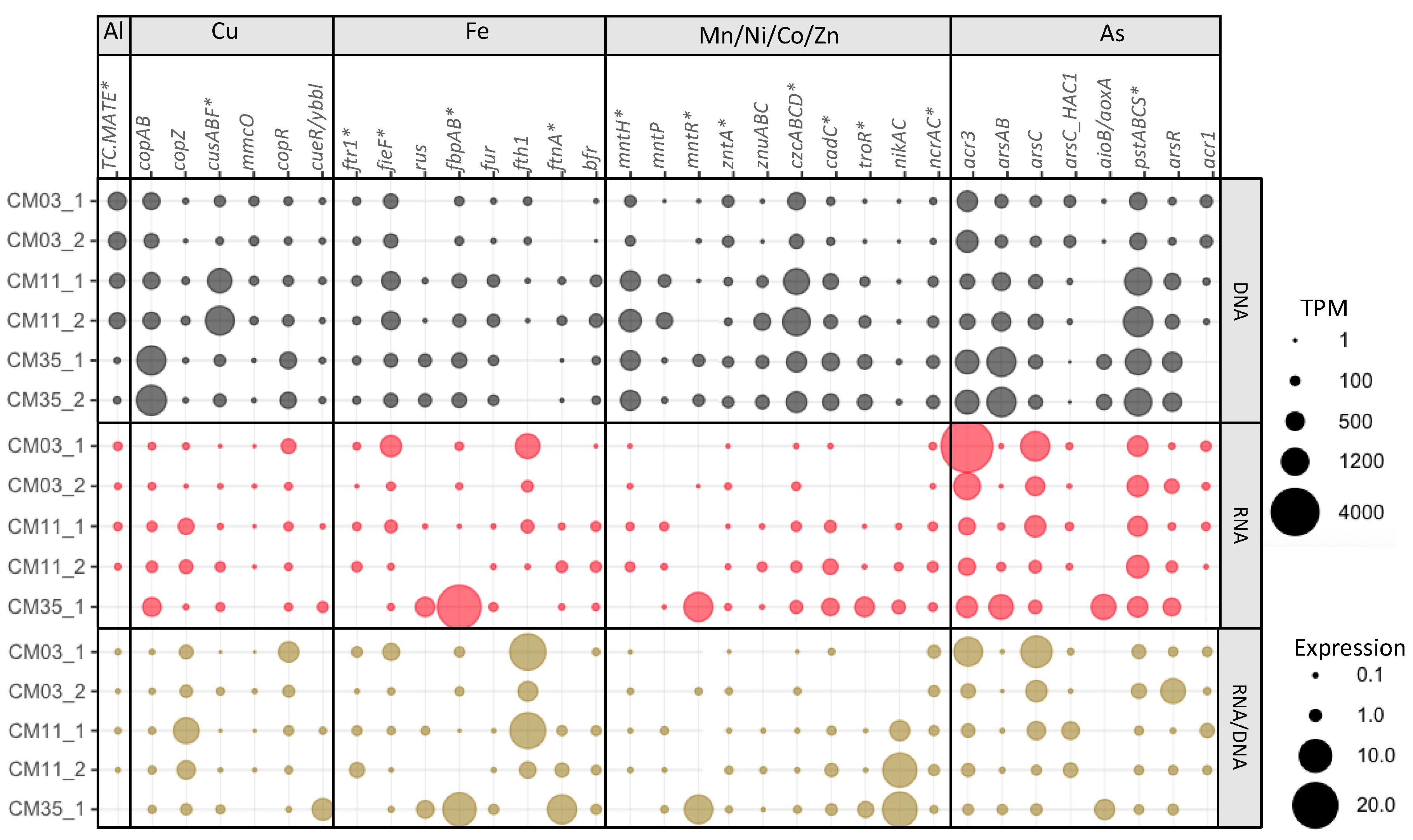
3.4. Element-Specific Response Mechanisms
3.4.1. Aluminum
3.4.2. Copper
3.4.3. Iron
3.4.4. Manganese, Nickel, Cobalt, and Zinc
3.4.5. Arsenic
3.4.6. Other Metal Response Mechanisms
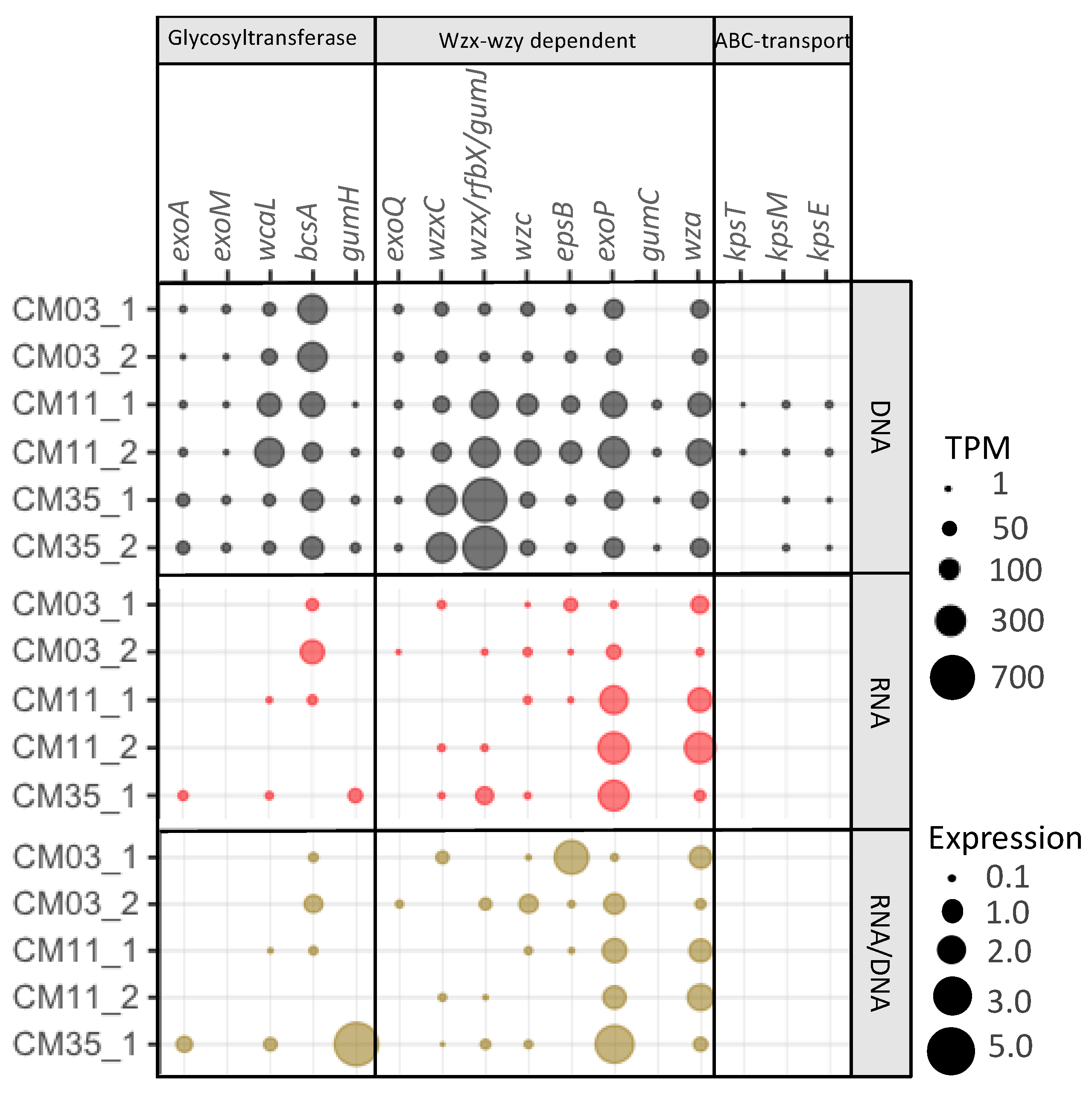
3.5. Extracellular Metal Sequestration
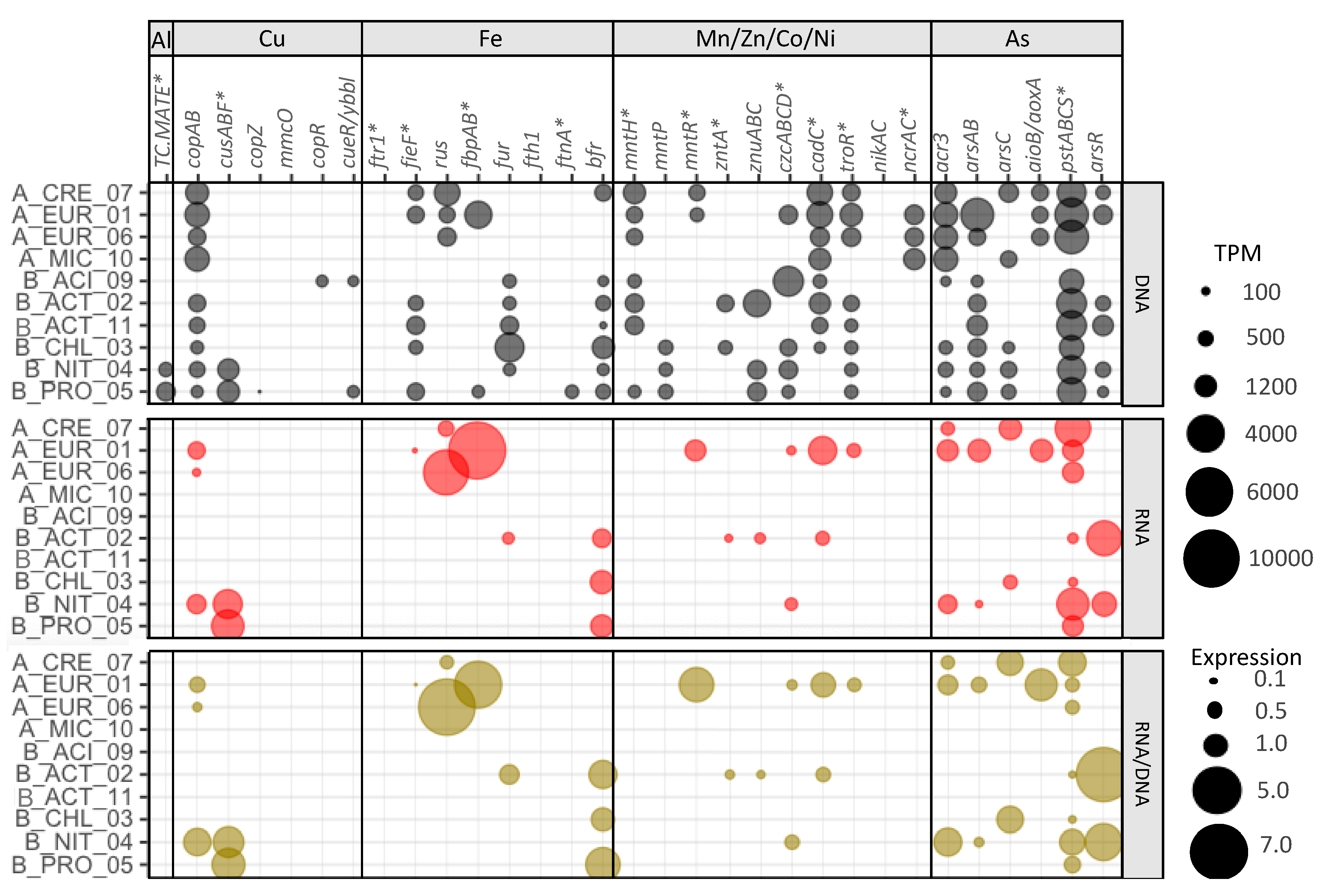
3.6. Metal Resistance Mechanisms in Deep Layer Populations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrer, A.; Orellana, O.; Levicán, G. Oxidative Stress and Metal Tolerance in Extreme Acidophiles. In Acidophiles, Life in Extremely Acidic Environments; Quatrini, R., Johnson, B., Eds.; Caister Academic Press: Norfolk, UK, 2016; pp. 63–76. [Google Scholar]
- Slyemi, D.; Bonnefoy, V. How prokaryotes deal with arsenic. Environ. Microbiol. Rep. 2012, 4, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Ben Fekih, I.; Zhang, C.; Li, Y.P.; Zhao, Y.; Alwathnani, H.A.; Saquib, Q.; Rensing, C.; Cervantes, C. Distribution of Arsenic Resistance Genes in Prokaryotes. Front. Microbiol. 2018, 9, 2473. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Hu, Y.; Luo, J.; Xu, B.; Zhao, J. Geochemical processes controlling fate and transport of arsenic in acid mine drainage (AMD) and natural systems. J. Hazard. Mater. 2009, 165, 13–26. [Google Scholar] [CrossRef]
- Dopson, M.; Holmes, D.S. Metal resistance in acidophilic microorganisms and its significance for biotechnologies. Appl. Microbiol. Biotechnol. 2014, 98, 8133–8144. [Google Scholar] [CrossRef]
- Mangold, S.; Potrykus, J.; Björn, E.; Lövgren, L.; Dopson, M. Extreme zinc tolerance in acidophilic microorganisms from the bacterial and archaeal domains. Extremophiles 2013, 17, 75–85. [Google Scholar] [CrossRef]
- Navarro, C.A.; Orellana, L.H.; Mauriaca, C.; Jerez, C.A. Transcriptional and functional studies of Acidithiobacillus ferrooxidans genes related to survival in the presence of copper. Appl. Environ. Microbiol. 2009, 75, 6102–6109. [Google Scholar] [CrossRef]
- Osorio, H.; Martínez, V.; Nieto, P.A.; Holmes, D.S.; Quatrini, R. Microbial iron management mechanisms in extremely acidic environments: Comparative genomics evidence for diversity and versatility. BMC Microbiol. 2008, 8, 203. [Google Scholar] [CrossRef]
- Tian, J.; Wu, N.; Li, J.; Liu, Y.; Guo, J.; Yao, B.; Fan, Y. Nickel-resistant determinant from Leptospirillum ferriphilum. Appl. Environ. Microbiol. 2007, 73, 2364–2368. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Dopson, M.; Wexler, M.; Sawers, R.G.; Stemmler, A.; Rosen, B.P.; Bond, P.L. Extreme arsenic resistance by the acidophilic archaeon ‘Ferroplasma acidarmanus’ Fer1. Extremophiles 2007, 11, 425–434. [Google Scholar] [CrossRef]
- Hirooka, S.; Hirose, Y.; Kanesaki, Y.; Higuchi, S.; Fujiwara, T.; Onuma, R.; Era, A.; Ohbayashi, R.; Uzuka, A.; Nozaki, H. Acidophilic green algal genome provides insights into adaptation to an acidic environment. Proc. Natl. Acad. Sci. USA 2017, 114, E8304–E8313. [Google Scholar] [CrossRef] [PubMed]
- Koechler, S.; Bertin, P.N.; Plewniak, F.; Baltenweck, R.; Casiot, C.; Heipieper, H.J.; Bouchez, O.; Arsène-Ploetze, F.; Hugueney, P.; Halter, D. Arsenite response in Coccomyxa sp. Carn explored by transcriptomic and non-targeted metabolomic approaches. Environ. Microbiol. 2016, 18, 1289–1300. [Google Scholar] [CrossRef]
- Singh, A.P.; Goel, R.K.; Kaur, T. Mechanisms pertaining to arsenic toxicity. Toxicol. Int. 2011, 18, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Dopson, M.; Lindström, B.E.; Hallberg, K.B. Chromosomally encoded arsenical resistance of the moderately thermophilic acidophile Acidithiobacillus caldus. Extremophiles 2001, 5, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Tuffin, I.M.; Hector, S.B.; Deane, S.M.; Rawlings, D.E. Resistance determinants of a highly arsenic-resistant strain of Leptospirillum ferriphilum isolated from a commercial biooxidation tank. Appl. Environ. Microbiol. 2006, 72, 2247–2253. [Google Scholar] [CrossRef]
- Lieutaud, A.; Van Lis, R.; Duval, S.; Capowiez, L.; Muller, D.; Lebrun, R.; Lignon, S.; Fardeau, M.-L.; Lett, M.-C.; Nitschke, W. Arsenite oxidase from Ralstonia sp. 22 Characterization of the enzyme and its interaction with soluble cytochromes. J. Biol. Chem. 2010, 285, 20433–20441. [Google Scholar] [CrossRef] [PubMed]
- Van Lis, R.; Nitschke, W.; Duval, S.; Schoepp-Cothenet, B. Arsenics as bioenergetic substrates. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Retamal-Morales, G.; Tischler, D. Metal binding ability of microbial natural metal chelators and potential applications. Nat. Prod. Rep. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Diwan, B. Bacterial Exopolysaccharide mediated heavy metal removal: A Review on biosynthesis, mechanism and remediation strategies. Biotechnol. Rep. 2016, 13, 58–71. [Google Scholar] [CrossRef]
- Schmid, J.; Sieber, V.; Rehm, B. Bacterial exopolysaccharides: Biosynthesis pathways and engineering strategies. Front. Microbiol. 2015, 6, 496. [Google Scholar] [CrossRef]
- Oshima, T.; Kondo, K.; Ohto, K.; Inoue, K.; Baba, Y. Preparation of phosphorylated bacterial cellulose as an adsorbent for metal ions. React. Funct. Polym. 2008, 68, 376–383. [Google Scholar] [CrossRef]
- Cuthbertson, L.; Mainprize, I.L.; Naismith, J.H.; Whitfield, C. Pivotal roles of the outer membrane polysaccharide export and polysaccharide copolymerase protein families in export of extracellular polysaccharides in gram-negative bacteria. Microbiol. Mol. Biol. Rev. 2009, 73, 155–177. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta (BBA) Bioenerg. 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2013, 42, D737–D743. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Diez Ercilla, M.; Pérez Cerdán, F.; Yusta, I.; Boyce, A.J. Hydrological investigation of a multi-stratified pit lake using radioactive and stable isotopes combined with hydrometric monitoring. J. Hydrol. 2014, 511, 494–508. [Google Scholar] [CrossRef]
- Diez-Ercilla, M. Estudio Hidrogeoquímico del Lago ácido de Cueva de la Mora (IPB, Huelva): Controles Sobre la Concentración de Metales y Modelo de Estratificación. Ph.D Thesis, Universidad del Pais Vasco, Leioa, Spain, 2015. [Google Scholar]
- Sánchez-España, J.; Pamo, E.L.; Diez, M.; Santofimia, E. Physico-chemical gradients and meromictic stratification in Cueva de la Mora and other acidic pit lakes of the Iberian Pyrite Belt. Mine Water Environ. 2009, 28, 15–29. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; Diez-Ercilla, M. Schwertmannite and hydrobasaluminite: A re-evaluation of their solubility and control on the iron and aluminium concentration in acidic pit lakes. Appl. Geochem. 2011, 26, 1752–1774. [Google Scholar] [CrossRef]
- Diez-Ercilla, M.; Falagán, C.; Yusta, I.; Sánchez-España, J. Metal mobility and mineral transformations driven by bacterial activity in acidic pit lake sediments: Evidence from column experiments and sequential extraction. J. Soils Sediments 2019, 19, 1527–1542. [Google Scholar] [CrossRef]
- Wendt-Potthoff, K.; Koschorreck, M.; Diez Ercilla, M.; Sánchez España, J. Microbial activity and biogeochemical cycling in a nutrient-rich meromictic acid pit lake. Limnol. Ecol. Manag. Inland Waters 2012, 42, 175–188. [Google Scholar] [CrossRef]
- Falagán, C.; Sánchez-España, J.; Johnson, D.B. New insights into the biogeochemistry of extremely acidic environments revealed by a combined cultivation-based and culture-independent study of two stratified pit lakes. FEMS Microbiol. Ecol. 2014, 87, 231–243. [Google Scholar] [CrossRef]
- Pinedo Vara, I. Piritas de Huelva: Su Historia, Minería y Aprovechamiento; Summa: Madrid, Spain, 1963. [Google Scholar]
- Tornos, F. Environment of formation and styles of volcanogenic massive sulfides: The Iberian Pyrite Belt. Ore Geol. Rev. 2006, 28, 259–307. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Pamo, E.L.; Pastor, E.S.; Ercilla, M.D. The acidic mine pit lakes of the Iberian Pyrite Belt: An approach to their physical limnology and hydrogeochemistry. Appl. Geochem. 2008, 23, 1260–1287. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Nonpareil: A redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2013, 30, 629–635. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Miller, C.S.; Baker, B.J.; Thomas, B.C.; Singer, S.W.; Banfield, J.F. EMIRGE: Reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011, 12, R44. [Google Scholar] [CrossRef]
- Pruesse, E.; Glöckner, F.O.; Peplies, J. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- West, P.T.; Probst, A.J.; Grigoriev, I.V.; Thomas, B.C.; Banfield, J.F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 2018, 28, 569–580. [Google Scholar] [CrossRef]
- Levy Karin, E.; Mirdita, M.; Söding, J. MetaEuk—Sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 2020, 8, 48. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Dedupe.sh. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/dedupe-guide/ (accessed on 2 May 2020).
- BBmap. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/ (accessed on 2 May 2020).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. bioRxiv 2019, 602110. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.B.; Mota, R.; Santos, C.L.; De Philippis, R.; Tamagnini, P. Chapter Seven—Assembly and Export of Extracellular Polymeric Substances (EPS) in Cyanobacteria: A Phylogenomic Approach. In Advances in Botanical Research; Chauvat, F., Cassier-Chauvat, C., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 235–279. [Google Scholar]
- Cuthbertson, L.; Kos, V.; Whitfield, C. ABC transporters involved in export of cell surface glycoconjugates. Microbiol. Mol. Biol. Rev. MMBR 2010, 74, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.A.; von Bernath, D.; Jerez, C.A. Heavy Metal Resistance Strategies of Acidophilic Bacteria and Their Acquisition: Importance for Biomining and Bioremediation. Biol. Res. 2013, 46, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Hemme, C.L.; Deng, Y.; Gentry, T.J.; Fields, M.W.; Wu, L.; Barua, S.; Barry, K.; Tringe, S.G.; Watson, D.B.; He, Z.; et al. Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J. 2010, 4, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Yu, H.; He, Z.; Deng, Y.; Wu, L.; Van Nostrand, J.D.; Zhou, A.; Voordeckers, J.; Lee, Y.-J.; Qin, Y.; et al. GeoChip 4: A functional gene-array-based high-throughput environmental technology for microbial community analysis. Mol. Ecol. Resour. 2014, 14, 914–928. [Google Scholar] [CrossRef]
- Hua, Z.-S.; Han, Y.-J.; Chen, L.-X.; Liu, J.; Hu, M.; Li, S.-J.; Kuang, J.-L.; Chain, P.S.G.; Huang, L.-N.; Shu, W.-S. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 2015, 9, 1280–1294. [Google Scholar] [CrossRef]
- Srivastava, P.; Kowshik, M. Mechanisms of Metal Resistance and Homeostasis in Haloarchaea. Archaea 2013, 2013, 732864. [Google Scholar] [CrossRef]
- Dopson, M.; Ossandon, F.J.; Lövgren, L.; Holmes, D.S. Metal resistance or tolerance? Acidophiles confront high metal loads via both abiotic and biotic mechanisms. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Fuentes, J.L.; Huss, V.A.R.; Montero, Z.; Torronteras, R.; Cuaresma, M.; Garbayo, I.; Vílchez, C. Phylogenetic characterization and morphological and physiological aspects of a novel acidotolerant and halotolerant microalga Coccomyxa onubensis sp. nov. (Chlorophyta, Trebouxiophyceae). J. Appl. Phycol. 2016, 28, 3269–3279. [Google Scholar] [CrossRef]
- Kusakizako, T.; Miyauchi, H.; Ishitani, R.; Nureki, O. Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim. Biophys. Acta (BBA) Biomembr. 2019, 183154. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.A.; von Bernath, D.; Martínez-Bussenius, C.; Castillo, R.A.; Jerez, C.A. Cytoplasmic CopZ-Like Protein and Periplasmic Rusticyanin and AcoP Proteins as Possible Copper Resistance Determinants in Acidithiobacillus ferrooxidans ATCC 23270. Appl. Environ. Microbiol. 2016, 82, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Lee, D.W.; Joo, H.K.; Jeong, K.H.; Lee, J.Y. Structural analysis of the manganese transport regulator MntR from Bacillus halodurans in apo and manganese bound forms. PLoS ONE 2019, 14, e0224689. [Google Scholar] [CrossRef] [PubMed]
- Panyushkina, A.E.; Babenko, V.V.; Nikitina, A.S.; Selezneva, O.V.; Tsaplina, I.A.; Letarova, M.A.; Kostryukova, E.S.; Letarov, A.V. Sulfobacillus thermotolerans: New insights into resistance and metabolic capacities of acidophilic chemolithotrophs. Sci. Rep. 2019, 9, 15069. [Google Scholar] [CrossRef] [PubMed]
- Orell, A.; Navarro, C.A.; Arancibia, R.; Mobarec, J.C.; Jerez, C.A. Life in blue: Copper resistance mechanisms of bacteria and Archaea used in industrial biomining of minerals. Biotechnol. Adv. 2010, 28, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, V.; Holmes, D.S. Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments. Environ. Microbiol. 2012, 14. [Google Scholar] [CrossRef]
- Ohmura, N.; Sasaki, K.; Matsumoto, N.; Saiki, H. Anaerobic Respiration Using Fe (3+), S(0), and H(2) in the Chemolithoautotrophic Bacterium Acidithiobacillus ferrooxidans. J. Bacteriol. 2002, 184, 2081–2087. [Google Scholar] [CrossRef]
- Kucera, J.; Janiczek, O.; Smoldas, J.; Mandl, M. Proteins Binding to Immobilized Rusticyanin Detected by Affinity Chromatography. Solid State Phenom. 2017, 262, 344–349. [Google Scholar] [CrossRef]
- Blanc, G.; Agarkova, I.; Grimwood, J.; Kuo, A.; Brueggeman, A.; Dunigan, D.D.; Gurnon, J.; Ladunga, I.; Lindquist, E.; Lucas, S.; et al. The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol. 2012, 13, R39. [Google Scholar] [CrossRef]
- Rivera, M. Bacterioferritin: Structure, Dynamics, and Protein-Protein Interactions at Play in Iron Storage and Mobilization. Acc. Chem. Res. 2017, 50, 331–340. [Google Scholar] [CrossRef]
- Falagán, C.; Johnson, D.B. Acidithiobacillus ferriphilus sp. nov., a facultatively anaerobic iron- and sulfur-metabolizing extreme acidophile. Int. J. Syst. Evol. Microbiol. 2016, 66, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Falagán, C.; Moya-Beltrán, A.; Castro, M.; Quatrini, R.; Johnson, D.B. Acidithiobacillus sulfuriphilus sp. nov.: An extremely acidophilic sulfur-oxidizing chemolithotroph isolated from a neutral pH environment. Int. J. Syst. Evol. Microbiol. 2019, 69, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Sandoval, M.; Storz, G. The Escherichia coli MntR miniregulon includes genes encoding a small protein and an efflux pump required for manganese homeostasis. J. Bacteriol. 2011, 193, 5887–5897. [Google Scholar] [CrossRef] [PubMed]
- Falagán, C.; Johnson, D.B. The significance of pH in dictating the relative toxicities of chloride and copper to acidophilic bacteria. Res. Microbiol. 2018, 169, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Heijerick, D.G.; De Schamphelaere, K.A.C.; Janssen, C.R. Biotic ligand model development predicting Zn toxicity to the alga Pseudokirchneriella subcapitata: Possibilities and limitations. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2002, 133, 207–218. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Yusta, I.; Burgos, W.D. Geochemistry of dissolved aluminum at low pH: Hydrobasaluminite formation and interaction with trace metals, silica and microbial cells under anoxic conditions. Chem. Geol. 2016, 441, 124–137. [Google Scholar] [CrossRef]
- Sánchez-España, J.; Wang, K.; Falagán, C.; Yusta, I.; Burgos, W.D. Microbially mediated aluminosilicate formation in acidic anaerobic environments: A cell-scale chemical perspective. Geobiology 2018, 16, 88–103. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Potrykus, J.; Wexler, M.; Bond, P.L.; Dopson, M. Biofilm development in the extremely acidophilic archaeon ‘Ferroplasma acidarmanus’ Fer1. Extremophiles 2010, 14, 485–491. [Google Scholar] [CrossRef]
- Falagán, C. Geomicrobiology of Meromictic Metal-Mine Pit Lakes in the Iberian Pyrite Belt and Biotechnological Applications. Ph.D Thesis, Universidad del Pais Vasco, Leioa, Spain, 2015. [Google Scholar]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725. [Google Scholar] [CrossRef]
- Tam, R.; Saier, M.H., Jr. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 1993, 57, 320–346. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shi, W.; Rosen, B.P. The chromosomal arsR gene of Escherichia coli encodes a trans-acting metalloregulatory protein. J. Biol. Chem. 1996, 271, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, J.P.; Valdés, J.; Quatrini, R.; Duarte, F.; Holmes, D.S. Lessons from the genomes of extremely acidophilic bacteria and archaea with special emphasis on bioleaching microorganisms. Appl. Microbiol. Biotechnol. 2010, 88, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Feng, X.; Tao, J.; Ma, L.; Xiao, Y.; Liang, Y.; Liu, X.; Yin, H. Comparative Genomics of the Extreme Acidophile Acidithiobacillus thiooxidans Reveals Intraspecific Divergence and Niche Adaptation. Int. J. Mol. Sci. 2016, 17, 1355. [Google Scholar] [CrossRef]
- Maguire, F.; Jia, B.; Gray, K.; Lau, W.Y.V.; Beiko, R.G.; Brinkman, F.S.L. Metagenome-Assembled Genome Binning Methods Disproportionately Fail for Plasmids and Genomic Islands. bioRxiv 2020. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3-m Depth Upper Oxic Layer | 11-m Depth Chemocline | 35-m Depth Deep Anoxic Layer | In-Stream Standard ^ | |
|---|---|---|---|---|
| pH | 2.6 | 3.95 | 4.5 | 6.5–9.0 |
| ORP (mV) | 575 | 60 | 41 | not specified |
| SC (mS/cm) | 3.4 | 4.85 | 12.1 | not specified |
| T (°C) | 17 | 12.5 | 18.3 | not specified |
| SO4 | 2,500,000 | 3,900,000 | 12,100,000 | 250,000 |
| Cl | 15,000 | 14,000 | 22,000 | 230,000 |
| Al | 140,000 | 158,000 | 5090 | 87 |
| As(III) | -- | -- | 17,200 | 150 |
| As(V) | 100 | 502 | 0.13 | |
| Co | 2010 | 1310 | 2040 | 19 |
| Cu | 6010 | 60 | 50 | 9.0 |
| Fe(III) | 118,000 | -- | -- | 1000 |
| Fe(II) | -- | 951,000 | 6,310,000 | |
| Mn | 19,000 | 35,200 | 116,000 | 167 * |
| Ni | 443 | 655 | 917 | 52 |
| Zn | 13,000 | 35,200 | 109,000 | 120 |
| PO4-P | <50 | <50 | 3000 | not specified |
| NH4-N | 25 | 400 | 536 | not specified |
| 3-M Depth Upper Oxic Layer | 11-M Depth Chemocline | 35-M Depth Deep Anoxic Layer | |
|---|---|---|---|
| Toxicity potency factor (TPF-1) ranking based on total concentration | Al > Cu > Fe(III) ~ Mn ~ Zn ~ Co > Ni > As(V) | Al > Fe(II) > Zn > Mn > Co > Ni > Cu > As(V) | Fe(II) >> Zn > Mn > As(III) ~ Co > Al > Ni > Cu |
| Toxicity potency factor (TPF-2) ranking based on free cation activity * | Cu > Al > Mn > Co > Zn > Fe(III) ~ Ni | Fe(II) > Al > Mn > Zn > Co > Ni > Cu | Fe(II) >> Mn > Zn > Co > Ni > Al > Cu |
| Al | TPF-1 = 1600; | TPF-1 = 1800; | TPF-1 = 60; |
| TPF-2 = 190 | TPF-2 = 230 | TPF-2 = 4.0 | |
| AlSO4+ = 83%; | AlSO4+ = 78%; | AlSO4+ = 72%; | |
| Al3+ = 12% | Al3+ = 13% | Al3+ = 6% | |
| As(V) | TPF-1 = 0.7; | TPF-1 = 3.3; | -- |
| TPF-2 = n.a. | TPF-2 = n.a. | ||
| H2AsO4– = 89% | H2AsO4– = 9% | ||
| As(III) | -- | -- | TPF-1 = 110; |
| TPF-2 = n.a. | |||
| H3AsO3 = 100% | |||
| Co | TPF-1 = 106; | TPF-1 = 69; | TPF-1 = 107; |
| TPF-2 = 78 | TPF-2 = 50 | TPF-2 = 85 | |
| Co2+ = 74%; | Co2+ = 73%; | Co2+ = 80%; | |
| CoSO4 = 26% | CoSO4 = 27% | CoSO4 = 20% | |
| Cu | TPF-1 = 670; | TPF-1 = 6.7; | TPF-1 = 5.6; |
| TPF-2 = 440 | TPF-2 = 3.1 | TPF-2 = 2.5 | |
| Cu2+ = 66%; | Cu2+ = 47%; | Cu2+ = 45%; | |
| CuSO4 = 34% | CuSO4 = 53% | CuSO4 = 55% | |
| Fe(III) | TPF-1 = 120; | -- | -- |
| TPF-2 = 6.0 | |||
| FeSO4+ = 51.0%; | |||
| Fe3+ = 5.0% | |||
| Fe(II) | -- | TPF-1 = 950; | TPF-1 = 6300; |
| TPF-2 = 660 | TPF-2 = 4800 | ||
| Fe2+ = 69.1%; | Fe2+ = 76.4%; | ||
| FeSO4 = 30.5% | FeSO4 = 23.6% | ||
| Mn | TPF-1 = 110; | TPF-1 = 210; | TPF-1 = 700; |
| TPF-2 = 90 | TPF-2 = 160 | TPF-2 = 570 | |
| Mn2+ = 76.2%; | Mn2+ = 76.0%; | Mn2+ = 81.8%; | |
| MnSO4 = 23.4% | MnSO4 = 23.9% | MnSO4 = 18.2% | |
| Ni | TPF-1 = 9.0; | TPF-1 = 13; | TPF-1 = 18; |
| TPF-2 = 6.0 | TPF-2 = 5.0 | TPF-2 = 8.0 | |
| Ni2+ = 68.6%; | NiSO4 = 62.2%; | NiSO4 = 54.9%; | |
| NiSO4 = 31.1% | Ni2+ = 37.5% | Ni2+ = 44.3% | |
| Zn | TPF-1 = 110; | TPF-1 = 290; | TPF-1 = 910; |
| TPF-2 = 70 | TPF-2 = 150 | TPF-2 = 130 | |
| Zn2+ = 63.9%; | Zn2+ = 52.7%; | Zn(SO4)22− = 67.0%; | |
| ZnSO4 = 31.7% | ZnSO4 = 34.6% | Zn2+ = 14.0% |
| Metal | Name | Description-Function | Mechanism | KO Identifier |
|---|---|---|---|---|
| Al | * TC.MATE | Multidrug resistance protein, part of the multidrug and toxin extrusion (MATE) family, Al and other drugs tolerance, also known as SLC47A, mtdK, dinF b | Export | K03327 |
| Cu | copAB | P-type Cu+ transporters, also involved in resistance to sodium acetate and Ag in certain organisms a, catalyze the translocation of inorganic cations b, copA also known as ctpA and ATP7 b, copB also known as copA_3, copF_3, cadA b | Export | K17686, K01533 |
| cueR/ybbI | MerR family transcriptional regulator, Cu efflux regulator, also involved in resistance to hydrochloric acid (HCl) a | Regulation | K19591 | |
| mmcO | Multicopper oxidase, oxidize metal ions (Cu) with oxygen as acceptor, also known as copA_1, copA_2, cueO b | Bioch Trans | K22552 | |
| copR | Two-component system, OmpR family, copper resistance phosphate regulon response regulator CusR b | Regulation | K07665 | |
| * cusABF | Cu(I)/Ag(I) efflux system membrane proteins, also known as silABF b | Export | K07787, K07798, K07810 | |
| copZ | Copper chaperone ab | Export | K07213 | |
| Fe | * ftr1 | High affinity low-pH Fe(II) transporter b, also involved in Pb resistance a | Import | K07243 |
| * fieF | Ferrous-iron efflux pump b, also involved in efflux of Zn/Co/Cd/Ni a | Export | K13283 | |
| * fbpAB | Iron(III) transport system substrate-binding protein, ABC transporters, also known as afuAB b, also involved in Ga resistance a | Import | K02012, K02011 | |
| * rus | Rusticyanin, involved in Fe(II) oxidation, but potentially in copper resistance [68] | Bioch Trans | K18683 | |
| fur | Fur family transcriptional regulator, ferric uptake regulator, also known as zur and furB b | Regulation | K03711 | |
| fth1 | Ferritin heavy chain, iron storage mainly found in eukaryotes b | Int Accu | K00522 | |
| * ftnA | Ferritin, iron storage, also involved in resistance to Cu and Mn a. | Int Accu | K02217 | |
| bfr | Bacterioferritin, iron storage b | Int Accu | K03594 | |
| Mn | mntP | Manganese efflux protein ab | Export | K23242 |
| * mntH | Manganese transport protein involved in Mn, Zn, and Fe uptake a | Import | K03322 | |
| * mntR | DtxR family transcriptional regulator, manganese transport regulator b, responds to Mn(II), Fe(II), Zn(II), Cd(II), Co(II) [69] | Regulation | K11924 | |
| Zn | * zntA | Zn2 + /Cd2 + -exporting ATPase b, also involved in Co and Pb extrusion a | Export | K01534 |
| znuABC | Zinc transport system b | Import | K09815, K09816, K09817 | |
| * czcABCD | Cobalt-zinc-cadmium efflux system b, also involved in Ni and Co resistance a | Export | K15726, K15727, K15725, K16264 | |
| * cadC | ArsR family transcriptional regulator, responsive transcriptional repressor b, involved in Cd/Bi/Zn/Pb resistance a | Regulation | K21903 | |
| * troR | Mn-dependent transcriptional regulator b, involved in resistance to Zn/Mn/Fe and Hydrogen Peroxide a | Regulation | K03709 | |
| Ni | nikAC | Nickel transport system b | Import | K15584, K15586 |
| * ncrAC | Ni/Co transporters, involved also in Co/Cd/Zn/Fe, also known as nrsD/rcnA/yohM ab | Export | K07785, K08970 | |
| As | acr3 | Arsenite transporter b | Export | K03325 |
| arsAB | Arsenite/tail-anchored protein-transporting ATPase and pump membrane protein b | Export | K01551, K03893 | |
| arsC | Arsenate reductase (thioredoxin as acceptor) b | Bioch Trans | K03741 | |
| arsC_HAC1 | Arsenate reductase (glutathione or glutaredoxin as acceptor) b | Bioch Trans | K22547 | |
| aioB/aoxA | Arsenite oxidase small subunit b | Bioch Trans | K08355 | |
| * pstABCS | Phosphate transport system, As(V) uptake a | Bioch Trans | K02038, K02036, K02037, K02040 | |
| arsR | Transcriptional repressor, As resistance, regulation b | Regulation | K03892 | |
| acr1 | AP-1-like transcription factor, As resistance, regulation b | Regulation | K09043 |
| Gene | Description-Function | Pathway | KO Identifier |
|---|---|---|---|
| exoQ | Exopolysaccharide production protein ExoQ (polymerase wzy) | Wzx-Wzy dependent pathway | K16567 |
| wzxC | wzxC; lipopolysaccharide exporter (flippase wzx) | Wzx-Wzy dependent pathway | K16695 |
| wzx/rfbX/gumJ | Polysaccharide transporter, PST family (flippase wzx) | Wzx-Wzy dependent pathway | K03328 |
| wzc | Tyrosine-protein kinase Etk/Wzc (polysaccharide co-polymerase PCP) | Wzx-Wzy dependent pathway | K16692 |
| epsB | Protein-tyrosine kinase (polysaccharide co-polymerase PCP) | Wzx-Wzy dependent pathway | K00903 |
| exoP | Polysaccharide biosynthesis transport protein (polysaccharide co-polymerase PCP) | Wzx-Wzy dependent pathway | K16554 |
| gumC | GumC protein (polysaccharide co-polymerase PCP) | Wzx-Wzy dependent pathway | K13661 |
| wza | Polysaccharide biosynthesis/export protein (outer membrane transporter OPX) | Wzx-Wzy dependent pathway | K01991 |
| kpsT | Capsular polysaccharide transport system ATP-binding protein (ABC-transporter) | ABC-transport | K09689 |
| kpsM | Capsular polysaccharide transport system permease protein (ABC-transporter) | ABC-transport | K09688 |
| kpsE | Capsular polysaccharide transport system permease protein (polysaccharide co-polymerase PCP) | ABC-transport | K10107 |
| exoA | Succinoglycan biosynthesis protein ExoA | Glycosyltransferase | K16557 |
| exoM | Succinoglycan biosynthesis protein ExoM | Glycosyltransferase | K16556 |
| wcaL | Colanic acid/amylovoran biosynthesis glycosyltransferase | Glycosyltransferase | K16703 |
| gumH | Alpha-1,3-mannosyltransferase | Glycosyltransferase | K13657 |
| bcsA | BcsA; cellulose synthase (UDP-forming) | Glycosyltransferase | K00694 |
| MAG | Taxonomy Based on the Genome Taxonomy Database (GTDB) | Rel. Abu. (%) | Com (%) | Con (%) |
|---|---|---|---|---|
| A_CRE_07 | d__Archaea;p__Crenarchaeota;c__Nitrososphaeria;o__Nitrososphaerales;f__UBA183;g__UBA183 | 0.7 | 95 | 5 |
| A_EUR_01 | d__Archaea;p__Thermoplasmatota;c__Thermoplasmata;o__Thermoplasmatales;f__GCA-001856825;g__GCA-001856825 | 10.3 | 94 | 3 |
| A_EUR_06 | d__Archaea;p__Thermoplasmatota;c__Thermoplasmata;o__Thermoplasmatales;f__Thermoplasmataceae | 1.2 | 94 | 1 |
| A_MIC_10 | d__Archaea;p__Micrarchaeota;c__Micrarchaeia;o__Micrarchaeales;f__Micrarchaeaceae;g__UBA12276 | 0.5 | 77 | 0 |
| A_NAN_12 | d__Archaea;p__Nanoarchaeota;c__Nanoarchaeia;o__Woesearchaeales;f__UBA525;g__UBA153 | 0.4 | 77 | 0 |
| B_ACI_09 | d__Bacteria;p__Acidobacteriota;c__Acidobacteriae | 0.5 | 81 | 4 |
| B_ACT_02 | d__Bacteria;p__Actinobacteriota;c__Thermoleophilia;o__BMS3ABIN01;f__BMS3ABIN01 | 2.8 | 87 | 1 |
| B_ACT_11 | d__Bacteria;p__Actinobacteriota;c__Thermoleophilia;o__BMS3ABIN01;f__BMS3ABIN01; | 0.4 | 90 | 3 |
| B_CHL_03 | d__Bacteria;p__Chloroflexota;c__Dehalococcoidia;o__SZUA-161;f__SZUA-161 | 2.3 | 98 | 2 |
| B_DOR_08 | d__Bacteria;p__Dormibacterota;c__Dormibacteria;o__UBA8260;f__UBA8260 | 0.7 | 88 | 0 |
| B_NIT_04 | d__Bacteria;p__Nitrospirota;c__Thermodesulfovibrionia;o__Thermodesulfovibrionales;f__JdFR-88 | 2.3 | 100 | 1 |
| B_PAT_13 | d__Bacteria;p__Patescibacteria;c__Paceibacteria;o__UBA6257;f__Colwellbacteraceae | 0.2 | 75 | 0 |
| B_PRO_05 | d__Bacteria;p__Desulfobacterota;c__Desulfomonilia;o__Desulfomonilales;f__Desulfomonilaceae | 1.3 | 92 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayala-Muñoz, D.; Burgos, W.D.; Sánchez-España, J.; Couradeau, E.; Falagán, C.; Macalady, J.L. Metagenomic and Metatranscriptomic Study of Microbial Metal Resistance in an Acidic Pit Lake. Microorganisms 2020, 8, 1350. https://doi.org/10.3390/microorganisms8091350
Ayala-Muñoz D, Burgos WD, Sánchez-España J, Couradeau E, Falagán C, Macalady JL. Metagenomic and Metatranscriptomic Study of Microbial Metal Resistance in an Acidic Pit Lake. Microorganisms. 2020; 8(9):1350. https://doi.org/10.3390/microorganisms8091350
Chicago/Turabian StyleAyala-Muñoz, Diana, William D. Burgos, Javier Sánchez-España, Estelle Couradeau, Carmen Falagán, and Jennifer L. Macalady. 2020. "Metagenomic and Metatranscriptomic Study of Microbial Metal Resistance in an Acidic Pit Lake" Microorganisms 8, no. 9: 1350. https://doi.org/10.3390/microorganisms8091350
APA StyleAyala-Muñoz, D., Burgos, W. D., Sánchez-España, J., Couradeau, E., Falagán, C., & Macalady, J. L. (2020). Metagenomic and Metatranscriptomic Study of Microbial Metal Resistance in an Acidic Pit Lake. Microorganisms, 8(9), 1350. https://doi.org/10.3390/microorganisms8091350