Recognition of Candida albicans and Role of Innate Type 17 Immunity in Oral Candidiasis



{kind=link}
Abstract
1. Introduction
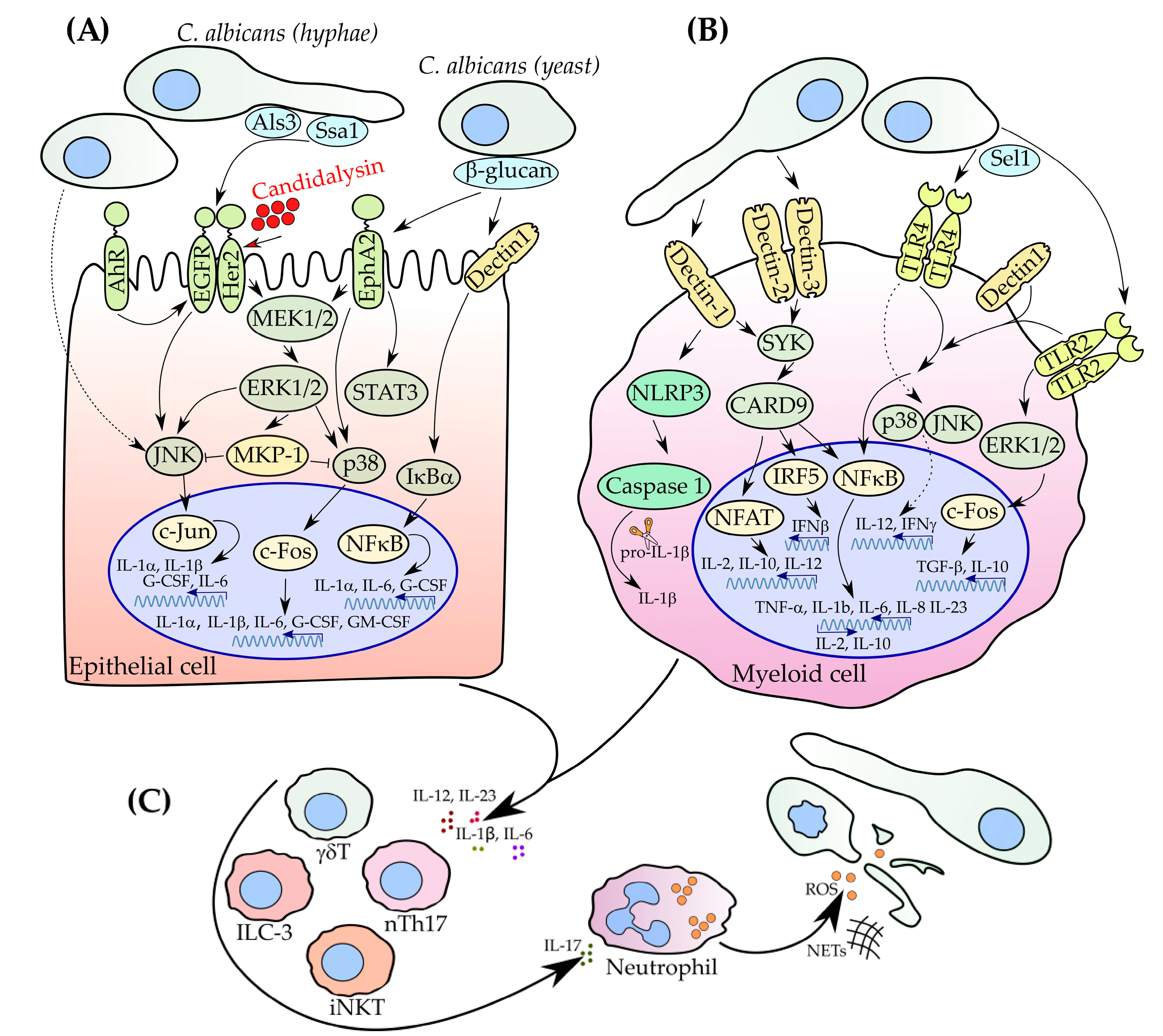
2. How Does C. albicans Invasion Trigger IL-17 Production?
2.1. Role of Myeloid Cells in Pathogen Recognition
2.2. Recognition of C. albicans by Epithelial Cells
3. IL-17 Orchestrates Innate and Adaptive Immunity
3.1. nTh17 Cells
3.2. γδ T Cells
3.3. iNKT
3.4. ILC3
4. Outlook and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Oever, J.T.; Netea, M.G. The bacteriome-mycobiome interaction and antifungal host defense. Eur. J. Immunol. 2014, 44, 3182–3191. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, M.; Ranjan, A.; Thompson, A.; Diaz, P.I.; Sobue, T.; Maas, K.; Dongari-Bagtzoglou, A. Candida albicans induces mucosal bacterial dysbiosis that promotes invasive infection. PLoS Pathog. 2019, 15, e1007717. [Google Scholar] [CrossRef] [PubMed]
- Bartnicka, D.; Gonzalez-Gonzalez, M.; Sykut, J.; Koziel, J.; Ciaston, I.; Adamowicz, K.; Bras, G.; Zawrotniak, M.; Karkowska-Kuleta, J.; Satala, D.; et al. Candida albicans Shields the Periodontal Killer Porphyromonas gingivalis from Recognition by the Host Immune System and Supports the Bacterial Infection of Gingival Tissue. Int. J. Mol. Sci. 2020, 21, 1984. [Google Scholar] [CrossRef] [PubMed]
- Passariello, C.; di Nardo, F.; Polimeni, A.; di Nardo, D.; Testarelli, L. Probiotic Streptococcus salivarius Reduces Symptoms of Denture Stomatitis and Oral Colonization by Candida albicans. Appl. Sci. 2020, 10, 3002. [Google Scholar] [CrossRef]
- Marol, S.; Yuecesoy, M. Molecular epidemiology of Candida species isolated from clinical specimens of intensive care unit patients. Mycoses 2008, 51, 40–49. [Google Scholar] [CrossRef]
- Eggimann, P.; Garbino, J.; Pittet, D. Epidemiology of Candida species infections in critically ill non-immunosuppressed patients. Lancet Infect. Dis. 2003, 3, 685–702. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Yadav, B. Microbe Profile: Candida albicans: A shape-changing, opportunistic pathogenic fungus of humans. Microbiol. SGM 2017, 163, 1145–1147. [Google Scholar] [CrossRef]
- Southern, P.; Horbul, J.; Maher, D.; Davis, D.A. C. albicans Colonization of Human Mucosal Surfaces. PLoS ONE 2008, 3, e2067. [Google Scholar] [CrossRef]
- Tsui, C.; Kong, E.F.; Jabra-Rizk, M.A. Pathogenesis of Candida albicans biofilm. Pathog. Dis. 2016, 74, ftw018. [Google Scholar] [CrossRef]
- Mukaremera, L.; Lee, K.K.; Mora-Montes, H.M.; Gow, N.A.R. Candida albicans Yeast, Pseudohyphal, and hyphal Morphogenesis Differentially affects immune recognition. Front. Immunol. 2017, 8, 629. [Google Scholar] [CrossRef]
- Boxx, G.M.; Kozel, T.R.; Nishiya, C.T.; Zhang, M.X. Influence of Mannan and Glucan on Complement Activation and C3 Binding by Candida albicans. Infect. Immun. 2010, 78, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M.; Louw, J.; Lewis, L.E.; Okai, B.; Walls, C.A.; Ballou, E.R.; Walker, L.A.; Reid, D.; Munro, C.A.; Brown, A.J.P.; et al. Candida albicans Hypha Formation and Mannan Masking of beta-Glucan Inhibit Macrophage Phagosome Maturation. Mbio 2014, 5, e01874. [Google Scholar] [CrossRef]
- Hofs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Bodey, G.P.; Mardani, M.; Hanna, H.A.; Boktour, M.; Abbas, J.; Girgawy, E.; Hachem, R.Y.; Kontoyiannis, D.P.; Raad, I.I. The epidemiology of Candida glabrata and Candida albicans fungemia in immunocompromised patients with cancer. Am. J. Med. 2002, 112, 380–385. [Google Scholar] [CrossRef]
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef]
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the Oral Fungal Microbiome (Mycobiome) in Healthy Individuals. PLoS Pathog. 2010, 6, e1000713. [Google Scholar] [CrossRef]
- Dupuy, A.K.; David, M.S.; Li, L.; Heider, T.N.; Peterson, J.D.; Montano, E.A.; Dongari-Bagtzoglou, A.; Diaz, P.I.; Strausbaugh, L.D. Redefining the Human Oral Mycobiome with Improved Practices in Amplicon-based Taxonomy: Discovery of Malassezia as a Prominent Commensal. PLoS ONE 2014, 9, e90899. [Google Scholar] [CrossRef]
- Metwalli, K.H.; Khan, S.A.; Krom, B.P.; Jabra-Rizk, M.A. Streptococcus mutans, Candida albicans, and the Human Mouth: A Sticky Situation. PLoS Pathog. 2013, 9, e1003616. [Google Scholar] [CrossRef]
- Patton, L.L. Progress in understanding oral health and HIV/AIDS. Oral Dis. 2014, 20, 223–225. [Google Scholar] [CrossRef]
- Patil, S.; Majumdar, B.; Sarode, S.C.; Sarode, G.S.; Awan, K.H. Oropharyngeal Candidosis in HIV-Infected Patients—An Update. Front. Microbiol. 2018, 9, 980. [Google Scholar] [CrossRef]
- UNAIDS. UNAIDS (Joint United Nations Programme on HIV/AIDS) Data 2019; UNAIDS: Geneva, Switzerland, 2019. [Google Scholar]
- Van de Veerdonk, F.L.; Kullberg, B.J.; Netea, M.G. Pathogenesis of invasive candidiasis. Curr. Opin. Crit. Care 2010, 16, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Hebecker, B.; Naglik, J.R.; Hube, B.; Jacobsen, I.D. Pathogenicity mechanisms and host response during oral Candida albicans infections. Expert Rev. Anti Infect. Ther. 2014, 12, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Joosten, L.A.B.; Kullberg, B.J.; Netea, M.G. Interplay between Candida albicans and the Mammalian Innate Host Defense. Infect. Immun. 2012, 80, 1304–1313. [Google Scholar] [CrossRef]
- Byrd, A.S.; O’Brien, X.M.; Johnson, C.M.; Lavigne, L.M.; Reichner, J.S. An Extracellular Matrix-Based Mechanism of Rapid Neutrophil Extracellular Trap Formation in Response to Candida albicans. J. Immunol. 2013, 190, 4136–4148. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef]
- Dutzan, N.; Kajikawa, T.; Abusleme, L.; Greenwell-Wild, T.; Zuazo, C.E.; Ikeuchi, T.; Brenchley, L.; Abe, T.; Hurabielle, C.; Martin, D.; et al. A dysbiotic microbiome triggers T(H)17 cells to mediate oral mucosal immunopathology in mice and humans. Sci. Transl. Med. 2018, 10, eaat0797. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Moutsopoulos, N.M. Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci. Immunol. 2020, 5, eaau4594. [Google Scholar] [CrossRef]
- Conti, H.R.; Peterson, A.C.; Brane, L.; Huppler, A.R.; Hernandez-Santos, N.; Whibley, N.; Garg, A.V.; Simpson-Abelson, M.R.; Gibson, G.A.; Mamo, A.J.; et al. Oral-resident natural Th17 cells and gamma delta T cells control opportunistic Candida albicans infections. J. Exp. Med. 2014, 211, 2075–2084. [Google Scholar] [CrossRef]
- Rosati, D.; Bruno, M.; Jaeger, M.; Oever, J.T.; Netea, M.G. Recurrent Vulvovaginal Candidiasis: An Immunological Perspective. Microorganisms 2020, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Mengesha, B.G.; Conti, H.R. The Role of IL-17 in Protection against Mucosal Candida Infections. J. Fungi 2017, 3, 52. [Google Scholar] [CrossRef]
- Peters, B.M.; Coleman, B.M.; Willems, H.M.E.; Barker, K.S.; Aggor, F.E.Y.; Cipolla, E.; Verma, A.H.; Bishu, S.; Huppler, A.H.; Bruno, V.M.; et al. The Interleukin (IL) 17R/IL-22R Signaling Axis Is Dispensable for Vulvovaginal Candidiasis Regardless of Estrogen Status. J. Infect. Dis. 2020, 221, 1554–1563. [Google Scholar] [CrossRef] [PubMed]
- Gow, N.A.R.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Gordon, S. Immune recognition of fungal beta-glucans. Cell. Microbiol. 2005, 7, 471–479. [Google Scholar] [CrossRef]
- Gow, N.A.R.; Latge, J.P.; Munro, C.A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Swidergall, M. Candida albicans at Host Barrier Sites: Pattern Recognition Receptors and Beyond. Pathogens 2019, 8, 40. [Google Scholar] [CrossRef]
- Pinheiro, C.R.; Coelho, A.L.; de Oliveira, C.E.; Gasparoto, T.H.; Garlet, G.P.; Silva, J.S.; Santos, C.F.; Cavassani, K.A.; Hogaboam, C.M.; Campanelli, A.P. Recognition of Candida albicans by gingival fibroblasts: The role of TLR2, TLR4/CD14, and MyD88. Cytokine 2018, 106, 67–75. [Google Scholar] [CrossRef]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A.R. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef]
- Swidergall, M.; Ernst, J.F. Interplay between Candida albicans and the Antimicrobial Peptide Armory. Eukaryot. Cell 2014, 13, 950–957. [Google Scholar] [CrossRef]
- Taylor, P.R.; Tsoni, S.V.; Willment, J.A.; Dennehy, K.M.; Rosas, M.; Findon, H.; Haynes, K.; Steele, C.; Botto, M.; Gordon, S.; et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 2007, 8, 31–38. [Google Scholar] [CrossRef]
- Zhu, L.L.; Zhao, X.Q.; Jiang, C.Y.; You, Y.; Chen, X.P.; Jiang, Y.Y.; Jia, X.M.; Lin, X. C-Type Lectin Receptors Dectin-3 and Dectin-2 Form a Heterodimeric Pattern-Recognition Receptor for Host Defense against Fungal Infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D.; Taylor, P.R.; Reid, D.M.; Willment, J.A.; Williams, D.L.; Martinez-Pomares, L.; Wong, S.Y.C.; Gordon, S. Dectin-1 is a major beta-glucan receptor on macrophages. J. Exp. Med. 2002, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.H.; et al. Dectin-2 Recognition of alpha-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.; Sousa, G.R.E. Myeloid C-type Lectin Receptors in Pathogen Recognition and Host Defense. Immunity 2011, 34, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.J.; Osorio, F.; Rosas, M.; Freitas, R.P.; Schweighoffer, E.; Gross, O.; SjefVerbeek, J.; Ruland, J.; Tybulewicz, V.; Brown, G.D.; et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 2009, 206, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Gewies, A.; Finger, K.; Schafer, M.; Sparwasser, T.; Peschel, C.; Forster, I.; Ruland, J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 2006, 442, 651–656. [Google Scholar] [CrossRef]
- Slack, E.C.; Robinson, M.J.; Hernanz-Falcon, P.; Brown, G.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.L.; Sousa, C.R.E. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur. J. Immunol. 2007, 37, 1600–1612. [Google Scholar] [CrossRef]
- Goodridge, H.S.; Simmons, R.M.; Underhill, D.M. Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J. Immunol. 2007, 178, 3107–3115. [Google Scholar] [CrossRef]
- Sancho, D.; Sousa, C.R.E. Signaling by Myeloid C-Type Lectin Receptors in Immunity and Homeostasis. Annu. Rev. Immunol. 2012, 30, 491–529. [Google Scholar] [CrossRef]
- Carvalho, A.; Giovannini, G.; de Luca, A.; D’Angelo, C.; Casagrande, A.; Iannitti, R.G.; Ricci, G.; Cunha, C.; Romani, L. Dectin-1 isoforms contribute to distinct Th1/Th17 cell activation in mucosal candidiasis. Cell. Mol. Immunol. 2012, 9, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Hohl, T.M.; van Epps, H.L.; Rivera, A.; Morgan, L.A.; Chen, P.L.; Feldmesser, M.; Pamer, E.G. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog. 2005, 1, 232–240. [Google Scholar] [CrossRef]
- Suram, S.; Brown, G.D.; Ghosh, M.; Gordon, S.; Loper, R.; Taylor, P.R.; Akira, S.; Uematsu, S.; Williams, D.L.; Leslie, C.C. Regulation of cytosolic phospholipase A(2) activation and cyclooxygenase 2 expression in macrophages by the beta-glucan receptor. J. Biol. Chem. 2006, 281, 5506–5514. [Google Scholar] [CrossRef] [PubMed]
- Suram, S.; Gangelhoff, T.A.; Taylor, P.R.; Rosas, M.; Brown, G.D.; Bonventre, J.V.; Akira, S.; Uematsu, S.; Williams, D.L.; Murphy, R.C.; et al. Pathways Regulating Cytosolic Phospholipase A(2) Activation and Eicosanoid Production in Macrophages by Candida albicans. J. Biol. Chem. 2010, 285, 30676–30685. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.G.; Padilla, J.; Koumas, L.; Ray, D.; Phipps, R.P. Prostaglandins as modulators of immunity. Trends Immunol. 2002, 23, 144–150. [Google Scholar] [CrossRef]
- Aoki, T.; Narumiya, S. Prostaglandins and chronic inflammation. Trends Pharmcol. Sci. 2012, 33, 304–311. [Google Scholar] [CrossRef]
- Noverr, M.C.; Phare, S.M.; Toews, G.B.; Coffey, M.J.; Huffnagle, G.B. Pathogenic yeasts Cryptococcus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect. Immun. 2001, 69, 2957–2963. [Google Scholar] [CrossRef]
- Tan, T.G.; Lim, Y.S.; Tan, A.; Leong, R.; Pavelka, N. Fungal Symbionts Produce Prostaglandin E-2 to Promote Their Intestinal Colonization. Front. Cell. Infect. Microbiol. 2019, 9, 359. [Google Scholar] [CrossRef]
- Marquez, S.; Fernandez, J.J.; Mancebo, C.; Herrero-Sanchez, C.; Alonso, S.; Sandoval, T.A.; Prados, M.R.; Cubillos-Ruiz, J.R.; Montero, O.; Fernandez, N.; et al. Tricarboxylic Acid Cycle Activity and Remodeling of Glycerophosphocholine Lipids Support Cytokine Induction in Response to Fungal Patterns. Cell Rep. 2019, 27, 525. [Google Scholar] [CrossRef]
- Strassmann, G.; Patilkoota, V.; Finkelman, F.; Fong, M.; Kambayashi, T. Evidence for the involvement of interleukin-10 in the differential deactivation of murine peritoneal-macrophages by prostaglandin-e(2). J. Exp. Med. 1994, 180, 2365–2370. [Google Scholar] [CrossRef]
- Liu, X.N.; Wang, D.; Yu, C.X.; Li, T.; Liu, J.Q.; Sun, S.J. Potential Antifungal Targets against a Candida Biofilm Based on an Enzyme in the Arachidonic Acid Cascade-A Review. Front. Microbiol. 2016, 7, 1925. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M.; Kullberg, B.J.; van de Veerdonk, F.L. Immune defence against Candida fungal infections. Nat. Rev. Immunol. 2015, 15, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Roeder, A.; Kirschning, C.J.; Schaller, M.; Weindl, G.; Wagner, H.; Korting, H.C.; Rupec, R.A. Induction of nuclear factor-kappa B and c-Jun/activator protein-1 via Toll-like receptor 2 in macrophages by antimycotic-treated Candida albicans. J. Infect. Dis. 2004, 190, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Murciano, C.; Yanez, A.; Gil, M.L.; Gozalbo, D. Both viable and killed Candida albicans cells induce in vitro production of TNF-alpha and IFN-gamma in murine cells through a TLR2-dependent signalling. Eur. Cytokine Netw. 2007, 18, 38–43. [Google Scholar] [PubMed]
- Archambault, L.S.; Trzilova, D.; Gonia, S.; Gale, C.; Wheeler, R.T. Intravital Imaging Reveals Divergent Cytokine and Cellular Immune Responses to Candida albicans and Candida parapsilosis. Mbio 2019, 10, e00266-19. [Google Scholar] [CrossRef]
- Gil, M.L.; Gozalbo, D. TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae. Microbes Infect. 2006, 8, 2299–2304. [Google Scholar] [CrossRef]
- Netea, M.G.; van der Meer, J.W.M.; Kullberg, B.J. Both TLR2 and TLR4 are involved in the recognition of Candida albicans. Reply to “TLR2, but not TLR4, triggers cytokine production by murine cells in response to Candida albicans yeasts and hyphae” by Gil and Gozalbo, Microbes and Infection. Microbes Infect. 2006, 8, 2821–2822. [Google Scholar] [CrossRef]
- Wang, W.J.; Deng, Z.H.; Wu, H.Y.; Zhao, Q.; Li, T.T.; Zhu, W.C.; Wang, X.J.; Tang, L.H.; Wang, C.S.; Cui, S.Z.; et al. A small secreted protein triggers a TLR2/4-dependent inflammatory response during invasive Candida albicans infection. Nat. Commun. 2019, 10, e00266-19. [Google Scholar] [CrossRef]
- Blasi, E.; Mucci, A.; Neglia, R.; Pezzini, F.; Colombari, B.; Radzioch, D.; Cossarizza, A.; Lugli, E.; Volpini, G.; Del Giudice, G.; et al. Biological importance of the two Toll-like receptors, TLR2 and TLR4, in macrophage response to infection with Candida albicans. Fems. Immunol. Med. Microbiol. 2005, 44, 69–79. [Google Scholar] [CrossRef]
- Hise, A.G.; Tomalka, J.; Ganesan, S.; Patel, K.; Hall, B.A.; Brown, G.D.; Fitzgerald, K.A. An Essential Role for the NLRP3 Inflammasome in Host Defense against the Human Fungal Pathogen Candida albicans. Cell Host Microbe 2009, 5, 487–497. [Google Scholar] [CrossRef]
- Gross, O.; Poeck, H.; Bscheider, M.; Dostert, C.; Hannesschlager, N.; Endres, S.; Hartmann, G.; Tardivel, A.; Schweighoffer, E.; Tybulewicz, V.; et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009, 459, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Rogiers, O.; Frising, U.C.; Kucharikova, S.; Jabra-Rizk, M.A.; van Loo, G.; van Dijck, P.; Wullaert, A. Candidalysin Crucially Contributes to Nlrp3 Inflammasome Activation by Candida albicans Hyphae. Mbio 2019, 10, e02221. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.; Konig, A.; Koenig, P.A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Gross, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.S.; Reading, P.C.; Jaillon, S.; Mantovani, A.; Mahalingam, S. Pentraxins and Collectins: Friend or Foe during Pathogen Invasion? Trends Microbiol. 2015, 23, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Harpf, V.; Rambach, G.; Wurzner, R.; Lass-Florl, C.; Speth, C. Candida and Complement: New Aspects in an Old Battle. Front. Immunol. 2020, 11, 1471. [Google Scholar] [CrossRef]
- Herrero-Sanchez, M.C.; Angomas, E.B.; de Ramon, C.; Telleria, J.J.; Corchete, L.A.; Alonso, S.; Ramos, M.D.; Penarrubia, M.J.; Marquez, S.; Fernandez, N.; et al. Polymorphisms in Receptors Involved in Opsonic and Nonopsonic Phagocytosis, and Correlation with Risk of Infection in Oncohematology Patients. Infect. Immun. 2018, 86, e00709-18. [Google Scholar] [CrossRef]
- Lee, G.W.; Lee, T.H.; Vilcek, J. TSG-14, A tumor necrosis factor-inducible and il-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J. Immunol. 1993, 150, 1804–1812. [Google Scholar]
- Alles, V.V.; Bottazzi, B.; Peri, G.; Golay, J.; Introna, M.; Mantovani, A. Inducible expression of ptx3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood 1994, 84, 3483–3493. [Google Scholar] [CrossRef]
- Ma, Y.J.; Doni, A.; Skjoedt, M.O.; Honore, C.; Arendrup, M.; Mantovani, A.; Garred, P. Heterocomplexes of Mannose-binding Lectin and the Pentraxins PTX3 or Serum Amyloid P Component Trigger Cross-activation of the Complement System. J. Biol. Chem. 2011, 286, 3405–3417. [Google Scholar] [CrossRef]
- Ma, Y.J.; Doni, A.; Hummelshoj, T.; Honore, C.; Bastone, A.; Mantovani, A.; Thielens, N.M.; Garred, P. Synergy between Ficolin-2 and Pentraxin 3 Boosts Innate Immune Recognition and Complement Deposition. J. Biol. Chem. 2009, 284, 28263–28275. [Google Scholar] [CrossRef]
- Jaeger, M.; van der Lee, R.; Cheng, S.C.; Johnson, M.D.; Kumar, V.; Ng, A.; Plantinga, T.S.; Smeekens, S.P.; Oosting, M.; Wang, X.; et al. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Bishu, S.; Hernandez-Santos, N.; Simpson-Abelson, M.R.; Huppler, A.R.; Conti, H.R.; Ghilardi, N.; Mamo, A.J.; Gaffen, S.L. The Adaptor CARD9 Is Required for Adaptive but Not Innate Immunity to Oral Mucosal Candida albicans Infections. Infect. Immun. 2014, 82, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.H.; Richardson, J.P.; Zhou, C.S.; Coleman, B.M.; Moyes, D.L.; Ho, J.; Huppler, A.R.; Ramani, K.; McGeachy, M.J.; Mufazalov, I.A.; et al. Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol. 2017, 2, eaam8834. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Runglall, M.; Murciano, C.; Shen, C.G.; Nayar, D.; Thavaraj, S.; Kohli, A.; Islam, A.; Mora-Montes, H.; Challacombe, S.J.; et al. A Biphasic Innate Immune MAPK Response Discriminates between the Yeast and Hyphal Forms of Candida albicans in Epithelial Cells. Cell Host Microbe 2010, 8, 225–235. [Google Scholar] [CrossRef]
- Zhu, W.D.; Phan, Q.T.; Boontheung, P.; Solis, N.V.; Loo, J.A.; Filler, S.G. EGFR and HER2 receptor kinase signaling mediate epithelial cell invasion by Candida albicans during oropharyngeal infection. Proc. Natl. Acad. Sci. USA 2012, 109, 14194–14199. [Google Scholar] [CrossRef]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64. [Google Scholar] [CrossRef]
- Solis, N.V.; Swidergall, M.; Bruno, V.M.; Gaffen, S.L.; Filler, S.G. The Aryl Hydrocarbon Receptor Governs Epithelial Cell Invasion during Oropharyngeal Candidiasis. Mbio 2017, 8, e00025-17. [Google Scholar] [CrossRef]
- Boutros, T.; Nantel, A.; Emadali, A.; Tzimas, G.; Conzen, S.; Chevet, E.; Metrakos, P.P. The MAP Kinase Phosphatase-1 MKP-1/DUSP1 Is a Regulator of Human Liver Response to Transplantation. Am. J. Transplant. 2008, 8, 2558–2568. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.H.; Zafar, H.; Ponde, N.O.; Hepworth, O.W.; Sihra, D.; Aggor, F.E.Y.; Ainscough, J.S.; Ho, J.; Richardson, J.P.; Coleman, B.M.; et al. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J. Immunol. 2018, 201, 627–634. [Google Scholar] [CrossRef]
- Swidergall, M.; Solis, N.V.; Lionakis, M.S.; Filler, S.G. EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat. Microbiol. 2018, 3, 53–61. [Google Scholar] [CrossRef]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc. Natl. Acad. Sci. USA 2000, 97, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Starnes, T.; Broxmeyer, H.E.; Robertson, M.J.; Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 2002, 169, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Z.B.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17—A novel cytokine derived from t-cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar]
- Lee, J.; Ho, W.H.; Maruoka, M.; Corpuz, R.T.; Baldwin, D.T.; Foster, J.S.; Goddard, A.D.; Yansura, D.G.; Vandlen, R.L.; Wood, W.I.; et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J. Biol. Chem. 2001, 276, 1660–1664. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.P.; Risser, P.; Maruoka, M.; Mao, W.G.; Foster, J.; Kelley, R.F.; et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef]
- Ferreira, M.C.; Whibley, N.; Mamo, A.J.; Siebenlist, U.; Chan, Y.R.; Gaffen, S.L. Interleukin-17-Induced Protein Lipocalin 2 Is Dispensable for Immunity to Oral Candidiasis. Infect. Immun. 2014, 82, 1030–1035. [Google Scholar] [CrossRef]
- Sanal, O.; Jing, H.E.; Ozgur, T.; Ayvaz, D.; Strauss-Albee, D.M.; Ersoy-Evans, S.; Tezcan, I.; Turkkani, G.; Matthews, H.F.; Haliloglu, G.; et al. Additional Diverse Findings Expand the Clinical Presentation of DOCK8 Deficiency. J. Clin. Immunol. 2012, 32, 698–708. [Google Scholar] [CrossRef]
- Edgerton, M.; Koshlukova, S.E.; Araujo, M.W.B.; Patel, R.C.; Dong, J.; Bruenn, J.A. Salivary histatin 5 and human neutrophil defensin 1 kill Candida albicans via shared pathways. Antimicrob. Agents Chemother. 2000, 44, 3310–3316. [Google Scholar] [CrossRef]
- Huppler, A.R.; Conti, H.R.; Hernandez-Santos, N.; Darville, T.; Biswas, P.S.; Gaffen, S.L. Role of Neutrophils in IL-17-Dependent Immunity to Mucosal Candidiasis. J. Immunol. 2014, 192, 1745–1752. [Google Scholar] [CrossRef]
- Trautwein-Weidner, K.; Gladiator, A.; Nur, S.; Diethelm, P.; LeibundGut-Landmann, S. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015, 8, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.R.; Nowyhed, H.N.; Choi, J.Y.; Poholek, A.C.; Odegard, J.M.; Flavell, R.A.; Craft, J. Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat. Immunol. 2009, 10, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, L.A.; Jain, R.; Haines, C.; Cua, D.J. Th17 cell development: From the cradle to the grave. Immunol. Rev. 2013, 252, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Yoshimoto, T.; Naka, T.; Nakae, S.; Iwakura, Y.; Cua, D.; Kubo, M. Natural Occurring IL-17 Producing T Cells Regulate the Initial Phase of Neutrophil Mediated Airway Responses. J. Immunol. 2009, 183, 7523–7530. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, W.E.; McCarthy, N.I.; Dutton, E.E.; Cowan, J.E.; Parnell, S.M.; White, A.J.; Anderson, G. Natural Th17 cells are critically regulated by functional medullary thymic microenvironments. J. Autoimmun. 2015, 63, 13–22. [Google Scholar] [CrossRef]
- Massot, B.; Michel, M.L.; Diem, S.; Ohnmacht, C.; Latour, S.; Dy, M.; Eberl, G.; Leite-de-Moraes, M.C. TLR-Induced Cytokines Promote Effective Proinflammatory Natural Th17 Cell Responses. J. Immunol. 2014, 192, 5635–5642. [Google Scholar] [CrossRef]
- Hernandez-Santos, N.; Wiesner, D.L.; Fites, J.S.; McDermott, A.J.; Warner, T.; Wuthrich, M.; Klein, B.S. Lung Epithelial Cells Coordinate Innate Lymphocytes and Immunity against Pulmonary Fungal Infection. Cell Host Microbe 2018, 23, 511. [Google Scholar] [CrossRef]
- Haas, W.; Pereira, P.; Tonegawa, S. GAMMA-DELTA Cells. Annu. Rev. Immunol. 1993, 11, 637–685. [Google Scholar] [CrossRef]
- Chien, Y.H.; Meyer, C.; Bonneville, M. Gamma delta T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of gamma delta T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef]
- Munoz-Ruiz, M.; Sumaria, N.; Pennington, D.J.; Silva-Santos, B. Thymic Determinants of gamma delta T Cell Differentiation. Trends Immunol. 2017, 38, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Hovav, A.H.; Wilharm, A.; Barel, O.; Prinz, I. Development and Function of gamma delta T Cells in the Oral Mucosa. J. Dent. Res. 2020, 99, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Wilharm, A.; Tabib, Y.; Nassar, M.; Reinhardt, A.; Mizraji, G.; Sandrock, I.; Heyman, O.; Barros-Martins, J.; Aizenbud, Y.; Khalaileh, A.; et al. Mutual interplay between IL-17-producing gamma delta T cells and microbiota orchestrates oral mucosal homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 2652–2661. [Google Scholar] [CrossRef]
- Fenoglio, D.; Poggi, A.; Catellani, S.; Battaglia, F.; Ferrera, A.; Setti, M.; Murdaca, G.; Zocchi, M.R. V delta 1 T lymphocytes producing IFN-gamma and IL-17 are expanded in HIV-1-infected patients and respond to Candida albicans. Blood 2009, 113, 6611–6618. [Google Scholar] [CrossRef]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans Stimulates IL-23 Release by Human Dendritic Cells and Downstream IL-17 Secretion by V delta 1 T Cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Itohara, S.; Farr, A.G.; Lafaille, J.J.; Bonneville, M.; Takagaki, Y.; Haas, W.; Tonegawa, S. Homing of a gamma-delta thymocyte subset with homogeneous t-cell receptors to mucosal epithelia. Nature 1990, 343, 754–757. [Google Scholar] [CrossRef]
- Prinz, I. Dynamics of the interaction of gamma delta T cells with their neighbors in vivo. Cell. Mol. Life Sci. 2011, 68, 2391–2398. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-Producing gamma delta T Cells Selectively Expand in Response to Pathogen Products and Environmental Signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H.G. Interleukin-1 and IL-23 Induce Innate IL-17 Production from gamma delta T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef]
- Monin, L.; Ushakov, D.S.; Arnesen, H.; Bah, N.; Jandke, A.; Munoz-Ruiz, M.; Carvalho, J.; Joseph, S.; Almeida, B.C.; Green, M.J.; et al. Gamma delta T cells compose a developmentally regulated intrauterine population and protect against vaginal candidiasis. Mucosal Immunol. 2020. [Google Scholar] [CrossRef]
- Ramani, K.; Jawale, C.V.; Verma, A.H.; Coleman, B.M.; Kolls, J.K.; Biswas, P.S. Unexpected kidney-restricted role for IL-17 receptor signaling in defense against systemic Candida albicans infection. JCI Insight 2018, 3, e98241. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Kaplan, D.H. Skin Immunity to Candida albicans. Trends Immunol. 2016, 37, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b(+) Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef]
- Rossjohn, J.; Pellicci, D.G.; Patel, O.; Gapin, L.; Godfrey, D.I. Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol. 2012, 12, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef]
- Coquet, J.M.; Chakravarti, S.; Kyparissoudis, K.; McNab, F.W.; Pitt, L.A.; McKenzie, B.S.; Berzins, S.P.; Smyth, M.J.; Godfrey, D.I. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4(−)NK1.1(−) NKT cell population. Proc. Natl. Acad. Sci. USA 2008, 105, 11287–11292. [Google Scholar] [CrossRef]
- Lee, Y.J.; Starrett, G.J.; Lee, S.T.; Yang, R.D.; Henzler, C.M.; Jameson, S.C.; Hogquist, K.A. Lineage-Specific Effector Signatures of Invariant NKT Cells Are Shared amongst gamma delta T, Innate Lymphoid, and Th Cells. J. Immunol. 2016, 197, 1460–1470. [Google Scholar] [CrossRef]
- Georgiev, H.; Ravens, I.; Benarafa, C.; Forster, R.; Bernhardt, G. Distinct gene expression patterns correlate with developmental and functional traits of iNKT subsets. Nat. Commun. 2016, 7, 13116. [Google Scholar] [CrossRef]
- Engel, I.; Seumois, G.; Chavez, L.; Samaniego-Castruita, D.; White, B.; Chawla, A.; Mock, D.; Vijayanand, P.; Kronenberg, M. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat. Immunol. 2016, 17, 728. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.T.; Benlagha, K.; Teyton, L.; Bendelac, A. Distinct functional lineages of human V alpha-24 natural killer T cells. J. Exp. Med. 2002, 195, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Teixeira, L.; Resende, M.; Coffre, M.; Devergne, O.; Herbeuval, J.P.; Hermine, O.; Schneider, E.; Rogge, L.; Ruemmele, F.M.; Dy, M.; et al. Proinflammatory Environment Dictates the IL-17-Producing Capacity of Human Invariant NKT Cells. J. Immunol. 2011, 186, 5758–5765. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.R.; Tatituri, R.V.V.; Rivera, A.; Watts, G.F.M.; Kim, E.Y.; Chiba, A.; Fuchs, B.B.; Mylonakis, E.; Besra, G.S.; Levitz, S.M.; et al. Innate Recognition of Cell Wall beta-Glucans Drives Invariant Natural Killer T Cell Responses against Fungi. Cell Host Microbe 2011, 10, 437–450. [Google Scholar] [CrossRef]
- Shimamura, M.; Yamamura, M.; Nabeshima, T.; Kitano, N.; van den Elzen, P.; Yesilkaya, H.; Andrew, P.; Illarionov, P. Activation of invariant natural killer T cells stimulated with microbial alpha-mannosyl glycolipids. Sci. Rep. 2017, 7, 9703. [Google Scholar] [CrossRef]
- Tarumoto, N.; Kinjo, Y.; Ueno, K.; Okawara, A.; Watarai, H.; Taniguchi, M.; Maesaki, S.; Miyazaki, Y. A Limited Role of iNKT Cells in Controlling Systemic Candida albicans Infections. Jpn. J. Infect. Dis. 2012, 65, 522–526. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; di Santo, J.P.; McKenzie, A.N.J. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting Edge: IL-17-Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef]
- Sparber, F.; Dolowschiak, T.; Mertens, S.; Lauener, L.; Clausen, B.E.; Joller, N.; Stoitzner, P.; Tussiwand, R.; LeibundGut-Landmann, S. Langerin(+) DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog. 2018, 14, e1007069. [Google Scholar] [CrossRef]
- Corte, L.; Roscini, L.; Colabella, C.; Tascini, C.; Leonildi, A.; Sozio, E.; Menichetti, F.; Merelli, M.; Scarparo, C.; Meyer, W.; et al. Exploring ecological modelling to investigate factors governing the colonization success in nosocomial environment of Candida albicans and other pathogenic yeasts. Sci. Rep. 2016, 6, 26860. [Google Scholar] [CrossRef] [PubMed]
- Cavalheiro, M.; Teixeira, M.C. Candida Biofilms: Threats, Challenges, and Promising Strategies. Front. Med. 2018, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Barata-Antunes, C.; Casal, M.; Brown, A.J.P.; van Dijck, P.; Paiva, S. Adapting to survive: How Candida overcomes host-imposed constraints during human colonization. PLoS Pathog. 2020, 16, e1008478. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.S.; Ozhegov, G.D.; Sabirova, A.E.; Novikova, V.V.; Lisovskaya, S.A.; Khabibrakhmanova, A.M.; Kurbangalieva, A.R.; Bogachev, M.I.; Kayumov, A.R. Increasing Susceptibility of Drug-Resistant Candida albicans to Fluconazole and Terbinafine by 2(5H)-Furanone Derivative. Molecules 2020, 25, 642. [Google Scholar] [CrossRef]
- Haqshenas, G.; Doerig, C. Targeting of host cell receptor tyrosine kinases by intracellular pathogens. Sci. Signal. 2019, 12, eaau9894. [Google Scholar] [CrossRef]
- Ho, J.; Moyes, D.L.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef]
- Vrijens, P.; Noppen, S.; Boogaerts, T.; Vanstreels, E.; Ronca, R.; Chiodelli, P.; Laporte, M.; Vanderinden, E.; Liekens, S.; Stevaert, A.; et al. Influenza virus entry via the GM3 ganglioside-mediated platelet-derived growth factor receptor beta signalling pathway. J. Gen. Virol. 2019, 100, 583–601. [Google Scholar] [CrossRef]
- Wu, S.Y.; Zhang, Q.; Zhang, F.; Meng, F.S.; Liu, S.D.; Zhou, R.Y.; Wu, Q.Z.; Li, X.R.; Shen, L.; Huang, J.; et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 2019, 21, 1027. [Google Scholar] [CrossRef]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Weinkauf, C.; Salvador, R.; PereiraPerrin, M. Neurotrophin Receptor TrkC Is an Entry Receptor for Trypanosoma cruzi in Neural, Glial, and Epithelial Cells. Infect. Immun. 2011, 79, 4081–4087. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. Cortactin: A Major Cellular Target of the Gastric Carcinogen Helicobacter pylori. Cancers 2020, 12, 159. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, M.; Thomsen, R.W.; Farkas, D.K.; Mogensen, M.F.; Sorensen, H.T. Candida infection and cancer risk: A Danish nationwide cohort study. Eur. J. Intern. Med. 2013, 24, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.M.; Liang, J.A.; Lin, C.L.; Sun, L.M.; Kao, C.H. Cancer risk in patients with candidiasis: A nationwide population-based cohort study. Oncotarget 2017, 8, 63562–63573. [Google Scholar] [CrossRef]
- Ramirez-Garcia, A.; Rementeria, A.; Aguirre-Urizar, J.M.; Moragues, M.D.; Antoran, A.; Pellon, A.; Abad-Diaz-de-Cerio, A.; Hernando, F.L. Candida albicans and cancer: Can this yeast induce cancer development or progression? Crit. Rev. Microbiol. 2016, 42, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Kazmierczak-Siedlecka, K.; Dvorak, A.; Folwarski, M.; Daca, A.; Przewlocka, K.; Makarewicz, W. Fungal Gut Microbiota Dysbiosis and Its Role in Colorectal, Oral, and Pancreatic Carcinogenesis. Cancers 2020, 12, 1326. [Google Scholar] [CrossRef]
- Tabarkiewicz, J.; Pogoda, K.; Karczmarczyk, A.; Pozarowski, P.; Giannopoulos, K. The Role of IL-17 and Th17 Lymphocytes in Autoimmune Diseases. Arch. Immunol. Et Ther. Exp. 2015, 63, 435–449. [Google Scholar] [CrossRef]
- Bernardini, N.; Skroza, N.; Tolino, E.; Mambrin, A.; Anzalone, A.; Balduzzi, V.; Colapietra, D.; Marchesiello, A.; Michelini, S.; Proietti, I.; et al. IL-17 and its role in inflammatory, autoimmune, and oncological skin diseases: State of art. Int. J. Dermatol. 2020, 59, 406–411. [Google Scholar] [CrossRef]
- Beringer, A.; Miossec, P. IL-17 and IL-17-producing cells and liver diseases, with focus on autoimmune liver diseases. Autoimmun. Rev. 2018, 17, 1176–1185. [Google Scholar] [CrossRef]
- Kuwabara, T.; Ishikawa, F.; Kondo, M.; Kakiuchi, T. The Role of IL-17 and Related Cytokines in Inflammatory Autoimmune Diseases. Mediat. Inflamm. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlova, A.; Sharafutdinov, I. Recognition of Candida albicans and Role of Innate Type 17 Immunity in Oral Candidiasis. Microorganisms 2020, 8, 1340. https://doi.org/10.3390/microorganisms8091340
Pavlova A, Sharafutdinov I. Recognition of Candida albicans and Role of Innate Type 17 Immunity in Oral Candidiasis. Microorganisms. 2020; 8(9):1340. https://doi.org/10.3390/microorganisms8091340
Chicago/Turabian StylePavlova, Anna, and Irshad Sharafutdinov. 2020. "Recognition of Candida albicans and Role of Innate Type 17 Immunity in Oral Candidiasis" Microorganisms 8, no. 9: 1340. https://doi.org/10.3390/microorganisms8091340
APA StylePavlova, A., & Sharafutdinov, I. (2020). Recognition of Candida albicans and Role of Innate Type 17 Immunity in Oral Candidiasis. Microorganisms, 8(9), 1340. https://doi.org/10.3390/microorganisms8091340