The Development of a Pipeline for the Identification and Validation of Small-Molecule RelA Inhibitors for Use as Anti-Biofilm Drugs

 ,
,  and
and 
Abstract
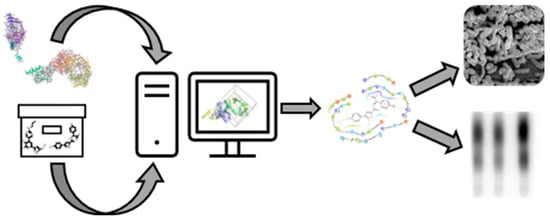
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Computational Docking
2.3. Biological Validation Assays
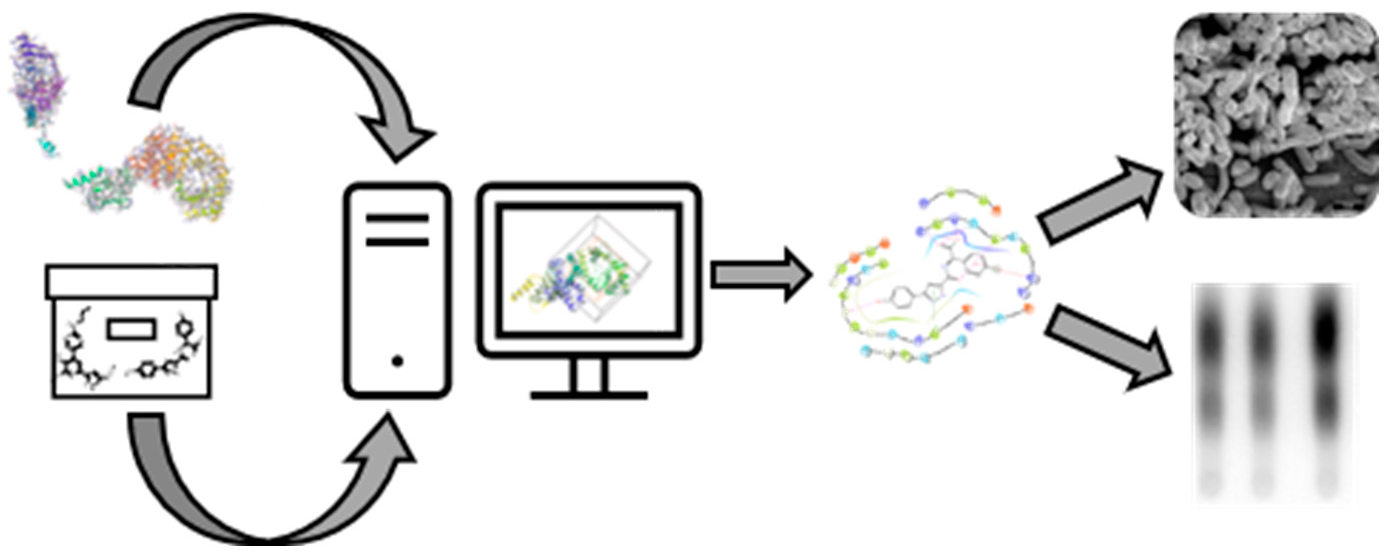
3. Results and Discussion
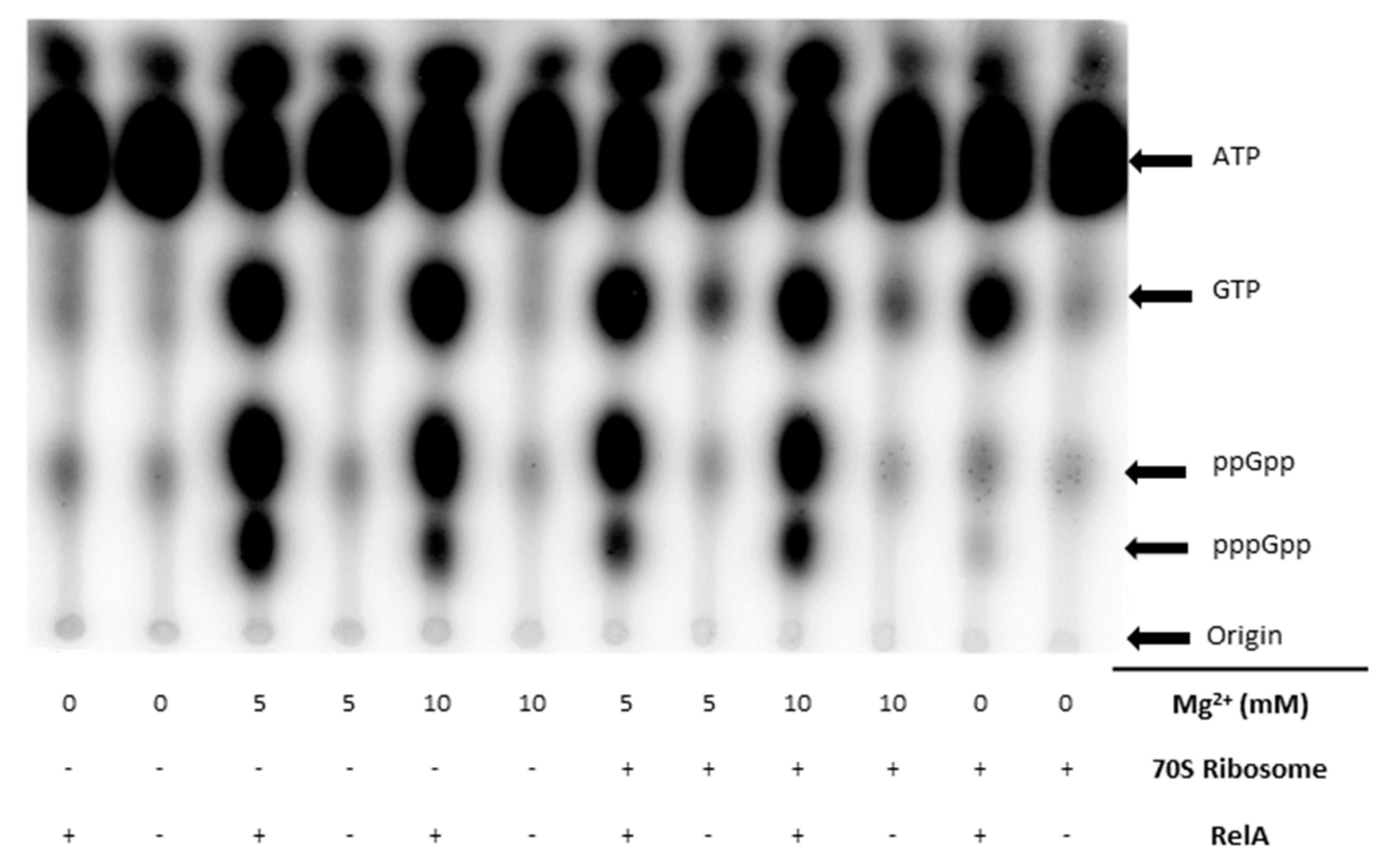
3.1. Validation of the RelA Activity Assays
3.2. Homology Studies
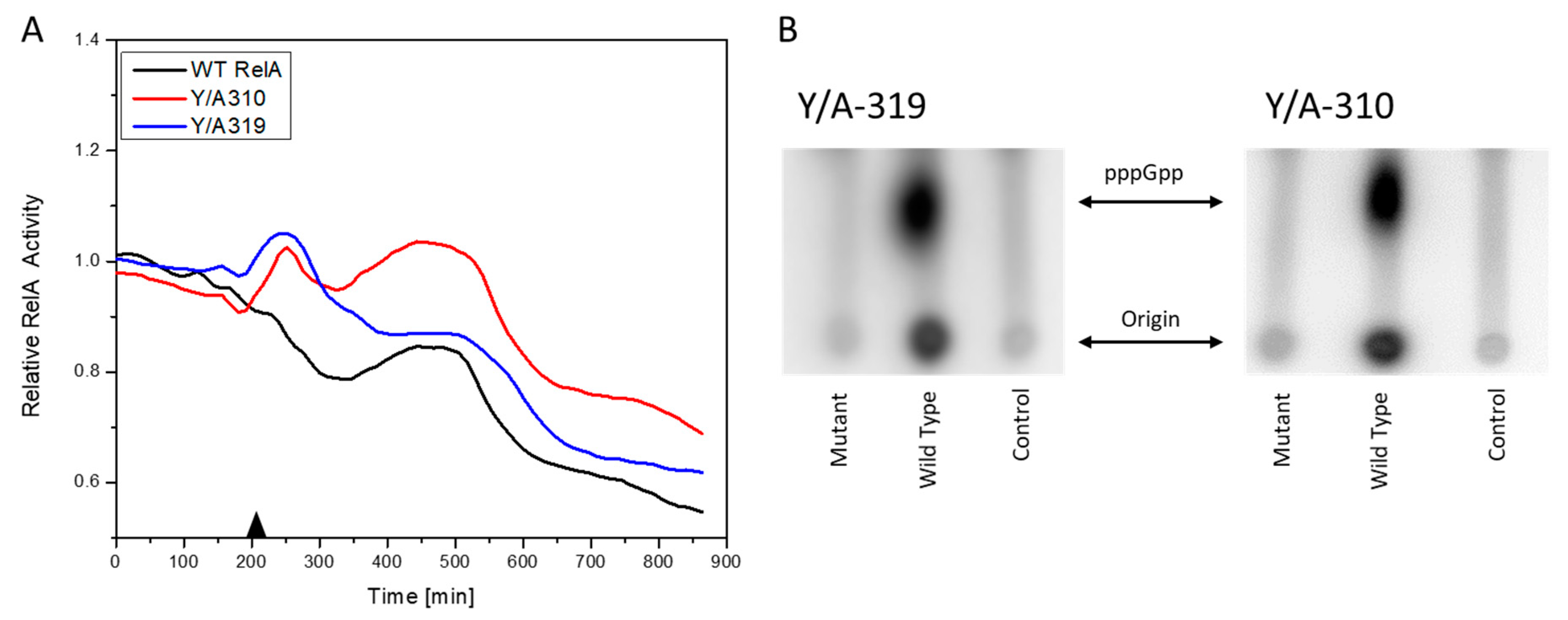
3.3. RelA Active Site Mutation Studies
3.4. In Silico Screening for Hit Compounds
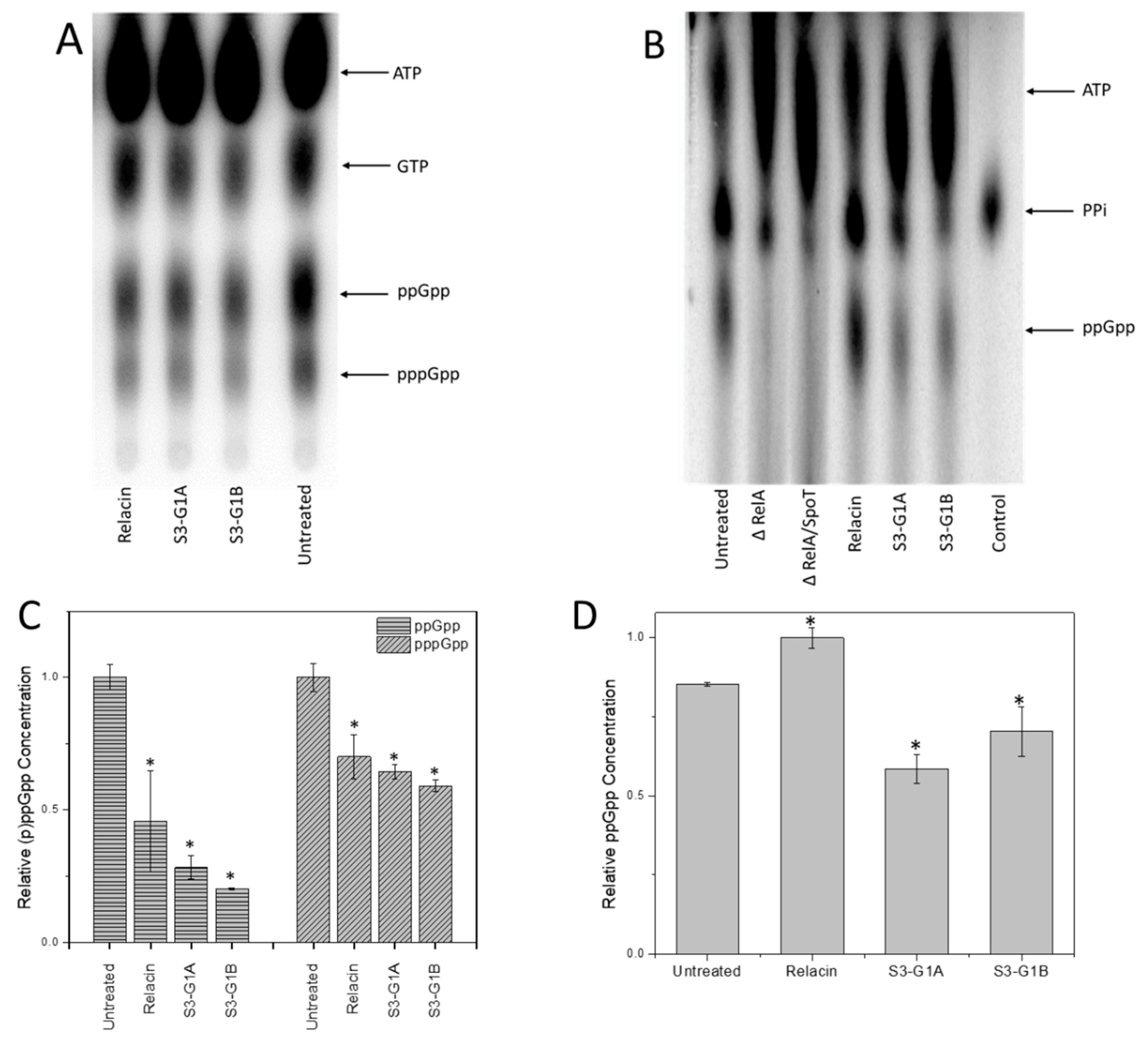
3.5. Effect of S3-G1A and S3-G1B on (p)ppGpp Production via In Vitro and In Vivo RelA Assays
3.6. Effect of Hit Compounds on Bacterial Growth
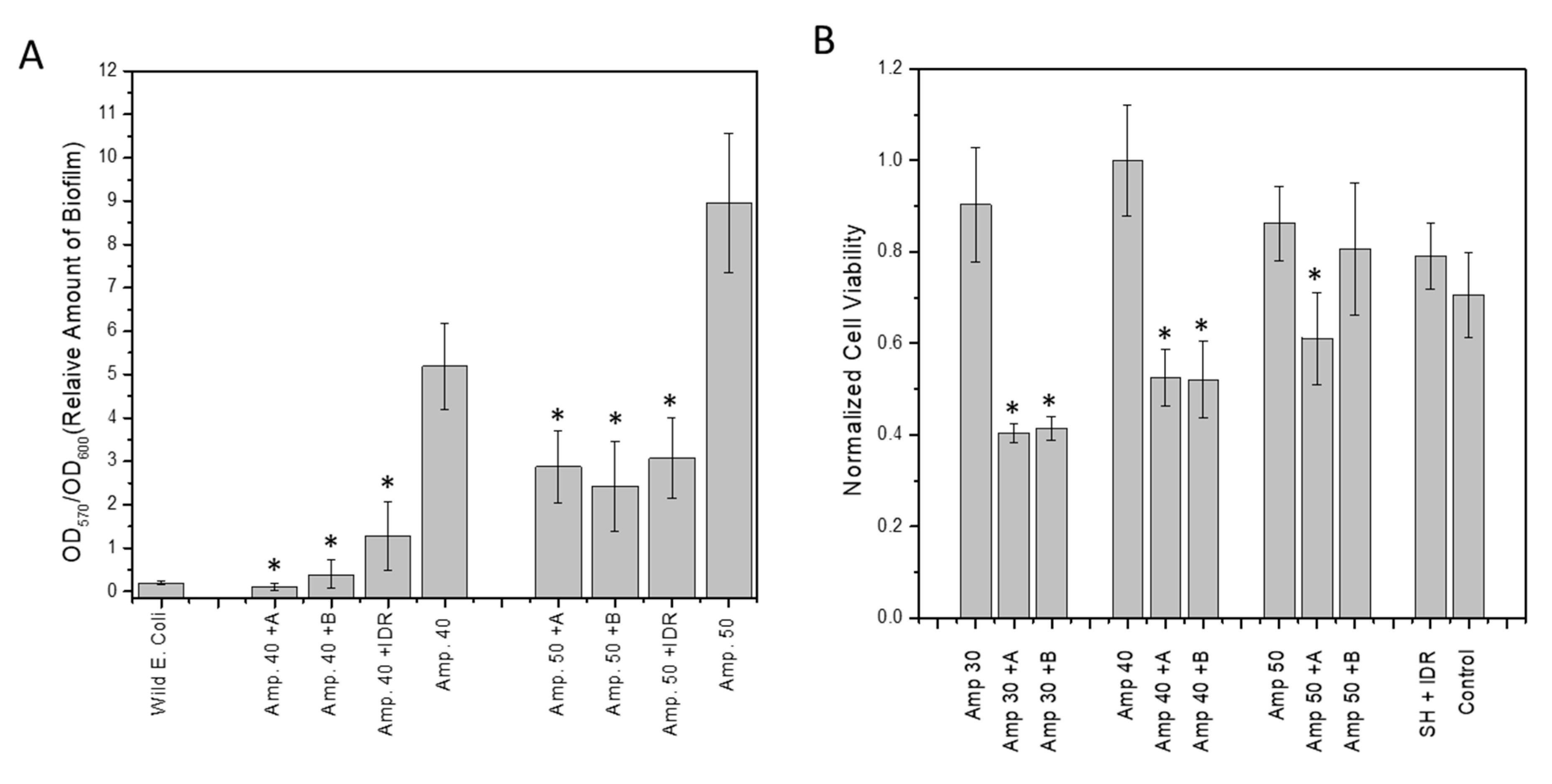
3.7. Effect of Hit Compounds on Biofilm Inhibition and Dispersal
3.8. Effect of Compound on Biofilm Persistence and Biofilm Viability
3.9. Effect of Hit Compounds on Biofilm Structure
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Materials and Correspondence
References
- Nadell, C.D.; Xavier, J.B.; Foster, K.R. The sociobiology of biofilms. FEMS Microbiol. Rev. 2008, 33, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Allegrucci, M.; Hu, F.Z.; Shen, K.; Hayes, J.; Ehrlich, G.D.; Post, J.C.; Sauer, K. Phenotypic characterization of Streptococcus pneumoniaebiofilm development. J. Bacteriol. 2006, 188. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, G.D.; Ahmed, A.; Earl, J.; Hiller, N.L.; Costerton, J.W.; Stoodley, P.; Post, J.C.; DeMeo, P.; Hu, F.Z. The distributed genome hypothesis as a rubric for understanding evolution in situ during chronic bacterial biofilm infectious processes. FEMS Immunol. Med. Microb. 2010, 59, 269–279. [Google Scholar] [CrossRef]
- Ehrlich, G.D.; Hiller, N.L.; Hu, F.Z. What makes pathogens pathogenic. Genome Boil. 2008, 9, 225. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, G.D.; Hu, F.Z.; Shen, K.; Stoodley, P.; Post, J.C. Bacterial plurality as a general mechanism driving persistence in chronic infections. Clin. Orthop. Relat. Res. 2005, 20–24. [Google Scholar] [CrossRef]
- Ito, A.; Taniuchi, A.; May, T.; Kawata, K.; Okabe, S. Increased Antibiotic Resistance of Escherichia coli in Mature Biofilms. Appl. Environ. Microbiol. 2009, 75, 4093–4100. [Google Scholar] [CrossRef]
- Costerton, W.; Veeh, R.; Shirtliff, M.; Pasmore, M.; Post, C.; Ehrlich, G. The application of biofilm science to the study and control of chronic bacterial infections. J. Clin. Investig. 2003, 112, 1466–1477. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Nistico, L.; Sambanthamoorthy, K.; Dice, B.; Nguyen, D.; Mershon, W.J.; Johnson, C.; Hu, F.Z.; Stoodley, P.; Ehrlich, G.D. Characterization of biofilm matrix, degradation by DNase treatment and evidence of capsule downregulation in Streptococcus pneumoniae clinical isolates. BMC Microbiol. 2008, 8, 173. [Google Scholar] [CrossRef]
- Janto, B.; Ahmed, A.; Ito, M.; Liu, J.; Hicks, D.B.; Pagni, S.; Fackelmayer, O.J.; Smith, T.-A.; Earl, J.; Elbourne, L.D.H.; et al. Genome of alkaliphilic Bacillus pseudofirmus OF4 reveals adaptations that support the ability to grow in an external pH range from 7.5 to 11.4. Environ. Microbiol. 2011, 13, 3289–3309. [Google Scholar] [CrossRef]
- Braxton, E.E., Jr.; Ehrlich, G.D.; Hall-Stoodley, L.; Stoodley, P.; Veeh, R.; Fux, C.; Hu, F.Z.; Quigley, M.; Post, J.C. Role of biofilms in neurosurgical device-related infections. Neurosurg. Rev. 2005, 28, 249–255. [Google Scholar] [CrossRef]
- Wolcott, R.D.; Ehrlich, G.D. Biofilms and Chronic Infections. JAMA 2008, 299, 2682–2684. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Cashel, M. The Control of Ribonucleic Acid Synthesis in Escherichia coli IV. Relevance of Unusual Phosphorylated Compounds from Amino Acid-Starved Stringent Strains. J. Biol. Chem. 1969, 244, 3133–3141. [Google Scholar] [PubMed]
- Haseltine, W.A.; Block, R. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc. Natl. Acad. Sci. USA 1973, 70, 1564–1568. [Google Scholar] [CrossRef]
- Cashel, M. Regulation of bacterial ppGpp and pppGpp. Ann. Rev. Microb. 1975, 29, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Nierlich, D.p. Regulation of bacterial growth. Science 1974, 184, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Erlich, H.; Laffler, T.; Gallant, J. ppGpp formation in Escherichia coli treated with rifampicin. J. Biol. Chem. 1971, 246, 6121–6123. [Google Scholar]
- Agirrezabala, X.; Fernández, I.S.; Kelley, A.C.; Cartón, D.G.; Ramakrishnan, V.; Valle, M. The ribosome triggers the stringent response by RelA via a highly distorted tRNA. Embo Rep. 2013, 14, 811–816. [Google Scholar] [CrossRef]
- Metzger, S.; Dror, I.B.; Aizenman, E.; Schreiber, G.; Toone, M.; Friesen, J.D.; Cashel, M.; Glaser, G. The nucleotide sequence and characterization of the relA gene of Escherichia coli. J. Biol. Chem. 1988, 263, 15699–15704. [Google Scholar]
- Loveland, A.B.; Bah, E.; Madireddy, R.; Zhang, Y.; Brilot, A.F.; Grigorieff, N.; Korostelev, A.A. Ribosome• RelA structures reveal the mechanism of stringent response activation. eLife 2016, 5, e17029. [Google Scholar] [CrossRef]
- Arenz, S.; Abdelshahid, M.; Sohmen, D.; Payoe, R.; Starosta, A.L.; Berninghausen, O.; Hauryliuk, V.; Beckmann, R.; Wilson, D.N. The stringent factor RelA adopts an open conformation on the ribosome to stimulate ppGpp synthesis. Nucleic Acids Res. 2016, 44, 6471–6481. [Google Scholar] [CrossRef] [PubMed]
- Gratani, F.L.; Horvatek, P.; Geiger, T.; Borisova, M.; Mayer, C.; Grin, I.; Wagner, S.; Steinchen, W.; Bange, G.; Velic, A. Regulation of the opposing (p) ppGpp synthetase and hydrolase activities in a bifunctional RelA/SpoT homologue from Staphylococcus aureus. PLoS Genet. 2018, 14, e1007514. [Google Scholar] [CrossRef] [PubMed]
- Ronneau, S.; Hallez, R. Make and break the alarmone: Regulation of (p) ppGpp synthetase/hydrolase enzymes in bacteria. FEMS Microbiol. Rev. 2019, 3, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Oppenheimer-Shaanan, Y.; Kaspy, I.; London, N.; Schueler-Furman, O.; Yavin, E.; Glaser, G.; Katzhendler, J.; Ben-Yehuda, S. Relacin, a Novel Antibacterial Agent Targeting the Stringent Response. PLoS Pathog. 2012, 8, e1002925. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Kaspy, I.; Glaser, G.; Katzhendler, J.; Yavin, E. Design, synthesis and structure–activity relationship of novel Relacin analogs as inhibitors of Rel proteins. Eur. J. Med. Chem. 2013, 70, 497–504. [Google Scholar] [CrossRef]
- Yanling, C.; Hongyan, L.; Xi, W.; Wim, C.; Dongmei, D. Efficacy of relacin combined with sodium hypochlorite against Enterococcus faecalis biofilms. J. Appl. Oral Sci. 2018, 26, e20160608. [Google Scholar] [CrossRef]
- Syal, K.; Flentie, K.; Bhardwaj, N.; Maiti, K.; Jayaraman, N.; Stallings, C.L.; Chatterji, D. Synthetic (p)ppGpp Analogue Is an Inhibitor of Stringent Response in Mycobacteria. Antimicrob. Agents Chemother. 2017, 61, e00443-17. [Google Scholar] [CrossRef]
- Dutta, N.K.; Klinkenberg, L.G.; Vazquez, M.-J.; Segura-Carro, D.; Colmenarejo, G.; Ramon, F.; Rodriguez-Miquel, B.; Mata-Cantero, L.; Porras-De Francisco, E.; Chuang, Y.-M.; et al. Inhibiting the stringent response blocks Mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci. Adv. 2019, 5, eaav2104. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-Spectrum Anti-biofilm Peptide That Targets a Cellular Stress Response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef]
- Pletzer, D.; Hancock, R.E. Antibiofilm Peptides: Potential as Broad-Spectrum Agents. J. Bacteriol. 2016, 198, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Dostert, M.; Belanger, C.R.; Hancock, R.E.W. Design and Assessment of Anti-Biofilm Peptides: Steps Toward Clinical Application. J. Innate Immun. 2019, 11, 193–204. [Google Scholar] [CrossRef]
- Mansour, S.C.; de la Fuente-Núñez, C.; Hancock, R.E.W. Peptide IDR-1018: Modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci. 2015, 21, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Andresen, L.; Tenson, T.; Hauryliuk, V. Cationic bactericidal peptide 1018 does not specifically target the stringent response alarmone (p)ppGpp. Sci. Rep. 2016, 6, 36549. [Google Scholar] [CrossRef]
- Wadood, A.; Ahmed, N.; Shah, L.; Ahmad, A.; Hassan, H.; Shams, S. In-silico drug design: An approach which revolutionarised the drug discovery process. OA Drug Des. Deliv. 2013, 1, 3–7. [Google Scholar]
- Brown, A.; Fernández, I.S.; Gordiyenko, Y.; Ramakrishnan, V. Ribosome-dependent activation of stringent control. Nature 2016, 534, 277–280. [Google Scholar] [CrossRef]
- Manav, M.C.; Beljantseva, J.; Bojer, M.S.; Tenson, T.; Ingmer, H.; Hauryliuk, V.; Brodersen, D.E. Structural basis for (p)ppGpp synthesis by the Staphylococcus aureus small alarmone synthetase RelP. J. Biol. Chem. 2018, 293, 3254–3264. [Google Scholar] [CrossRef]
- Steinchen, W.; Vogt, M.S.; Altegoer, F.; Giammarinaro, P.I.; Horvatek, P.; Wolz, C.; Bange, G. Structural and mechanistic divergence of the small (P)ppGpp synthetases RelP and RelQ. Sci. Rep. 2018, 8, 2195. [Google Scholar] [CrossRef]
- Hogg, T.; Mechold, U.; Malke, H.; Cashel, M.; Hilgenfeld, R. Conformational Antagonism between Opposing Active Sites in a Bifunctional RelA/SpoT Homolog Modulates (p)ppGpp Metabolism during the Stringent Response. Cell 2004, 117, 57–68. [Google Scholar] [CrossRef]
- Singal, B.; Balakrishna, A.M.; Nartey, W.; Manimekalai, M.S.S.; Jeyakanthan, J.; Gruber, G. Crystallographic and solution structure of the N-terminal domain of the Rel protein from Mycobacterium tuberculosis. FEBS Lett. 2017, 591, 2323–2337. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Shankarappa, B.; Sirko, D.; Ehrlich, G. A general method for the identification of regions suitable for site-directed silent mutagenesis. Biotechniques 1992, 12, 382–384. [Google Scholar] [PubMed]
- Shankarappa, B.; Vijayananda, K.; Ehrlich, G. SILMUT: A computer program for the identification of regions suitable for silent mutagenesis to introduce restriction enzyme recognition sequences. Biotechniques 1992, 12, 882–884. [Google Scholar]
- Shankarappa, B.; Balachandran, R.; Gupta, P.; Ehrlich, G. Introduction of multiple restriction enzyme sites by in vitro mutagenesis using the polymerase chain reaction. Genome Res. 1992, 1, 277–278. [Google Scholar] [CrossRef]
- Gawin, A.; Peebo, K.; Hans, S.; Ertesvåg, H.; Irla, M.; Neubauer, P.; Brautaset, T. Construction and characterization of broad-host-range reporter plasmid suitable for on-line analysis of bacterial host responses related to recombinant protein production. Microb. Cell Factories 2019, 18, 80. [Google Scholar] [CrossRef]
- Kitagawa, M.; Ara, T.; Arifuzzaman, M.; Ioka-Nakamichi, T.; Inamoto, E.; Toyonaga, H.; Mori, H. Complete set of ORF clones of Escherichia coli ASKA library (A Complete S et of E. coli K-12 ORF A rchive): Unique Resources for Biological Research. DNA Res. 2005, 12, 291–299. [Google Scholar] [CrossRef]
- Payoe, R.; Fahlman, R.P. Dependence of RelA-Mediated (p)ppGpp Formation on tRNA Identity. Biochemistry 2011, 50, 3075–3083. [Google Scholar] [CrossRef]
- Krol, J.E.; Biswas, S.; King, C.; Biswas, I. SMU.746-SMU.747, a putative membrane permease complex, is involved in aciduricity, acidogenesis, and biofilm formation in Streptococcus mutans. J. Bacteriol. 2014, 196, 129–139. [Google Scholar] [CrossRef]
- Król, J.E.; Hall, D.C.; Balashov, S.; Pastor, S.; Sibert, J.; McCaffrey, J.; Lang, S.; Ehrlich, R.L.; Earl, J.; Mell, J.C.; et al. Genome rearrangements induce biofilm formation in Escherichia coli C—An old model organism with a new application in biofilm research. BMC Genom. 2019, 20, 767. [Google Scholar] [CrossRef]
- Jaroslaw Krol, D.H.; Ehrlich, G. ASM Biofilms JEKmagTech 2018. In Proceedings of the ASM Biofilms 2018, Washington, DC, USA, 7–11 October 2018. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Aberg, A.; Fernández-Vázquez, J.; Cabrer-Panes, J.D.; Sánchez, A.; Balsalobre, C. Similar and divergent effects of ppGpp and DksA deficiencies on transcription in Escherichia coli. J. Bacteriol. 2009, 191, 3226–3236. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Olins, P.O.; Nomura, M. Regulation of the S10 ribosomal protein operon in E. coli: Nucleotide sequence at the start of the operon. Cell 1981, 26, 205–211. [Google Scholar] [CrossRef]
- Lemke, J.J.; Sanchez-Vazquez, P.; Burgos, H.L.; Hedberg, G.; Ross, W.; Gourse, R.L. Direct regulation of Escherichia coli ribosomal protein promoters by the transcription factors ppGpp and DksA. Proc. Natl. Acad. Sci. USA 2011, 108, 5712–5717. [Google Scholar] [CrossRef]
- Burgos, H.L.; O’Connor, K.; Sanchez-Vazquez, P.; Gourse, R.L. Roles of transcriptional and translational control mechanisms in regulation of ribosomal protein synthesis in Escherichia coli. J. Bact. 2017, 199, e00407–e00417. [Google Scholar] [CrossRef]
- Mechold, U.; Cashel, M.; Steiner, K.; Gentry, D.; Malke, H. Functional analysis of a relA/spoT gene homolog from Streptococcus equisimilis. J. Bacteriol. 1996, 178, 1401–1411. [Google Scholar] [CrossRef]
- Wendrich, T.M.; Blaha, G.; Wilson, D.N.; Marahiel, M.A.; Nierhaus, K.H. Dissection of the mechanism for the stringent factor RelA. Mol. Cell 2002, 10, 779–788. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Ji, H.-F.; Ehrlich, G.D.; Hall, D.C., Jr.; Krol, J.E.; Cahill, J.P. RelA Inhibitors for Biofilm Disruption. WO/2018/213185, 3 May 2020. [Google Scholar]
- Potrykus, K.; Murphy, H.; Philippe, N.; Cashel, M. ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 2011, 13, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Gaal, T.; Gourse, R.L. Guanosine 3′-diphosphate 5′-diphosphate is not required for growth rate-dependent control of rRNA synthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 5533–5537. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, V.J.; Bremer, H. Characterization of RNA and DNA synthesis in Escherichia coli strains devoid of ppGpp. J. Biol. Chem. 1993, 268, 10851–10862. [Google Scholar]
- Stoitsova, S.R.; Paunova-Krasteva, T.S.; Borisova, D.B. Modulation of biofilm growth by sub-inhibitory amounts of antibacterial substances. Microb. Biofilms-Importance Appl. 2016. [Google Scholar] [CrossRef]
- Bernier, S.P.; Lebeaux, D.; DeFrancesco, A.S.; Valomon, A.; Soubigou, G.; Coppée, J.-Y.; Ghigo, J.-M.; Beloin, C. Starvation, Together with the SOS Response, Mediates High Biofilm-Specific Tolerance to the Fluoroquinolone Ofloxacin. PLoS Genet. 2013, 9, e1003144. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Structure | IUPAC Name | Short Name | Binding Score (kcal/mol) | Difference from GTP (kcal/mol) |
|---|---|---|---|---|
 | (4-chlorophenyl)([(3-(4-hydroxyphenyl)-1H-pyrazol-5-yl]carbonyl)amino)acetic acid | S3-G1A | −10.38 | −1.71 |
 | 3-(6-amino-5-cyano-4-[2-(propylamino)pyrimidin-5-yl]pyridin-2-yl))-1H-pyrazole-5-carboxylic acid | S3-G1B | −9.64 | −0.97 |
 | Relacin | R | −8.67 | −0.08 |
 | Guanosine triphosphate | GTP | −8.67 | |
 | Adenosine triphosphate | ATP | −7.69 | 0.98 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, D.C., Jr.; Król, J.E.; Cahill, J.P.; Ji, H.-F.; Ehrlich, G.D. The Development of a Pipeline for the Identification and Validation of Small-Molecule RelA Inhibitors for Use as Anti-Biofilm Drugs. Microorganisms 2020, 8, 1310. https://doi.org/10.3390/microorganisms8091310
Hall DC Jr., Król JE, Cahill JP, Ji H-F, Ehrlich GD. The Development of a Pipeline for the Identification and Validation of Small-Molecule RelA Inhibitors for Use as Anti-Biofilm Drugs. Microorganisms. 2020; 8(9):1310. https://doi.org/10.3390/microorganisms8091310
Chicago/Turabian StyleHall, Donald C., Jr., Jarosław E. Król, John P. Cahill, Hai-Feng Ji, and Garth D. Ehrlich. 2020. "The Development of a Pipeline for the Identification and Validation of Small-Molecule RelA Inhibitors for Use as Anti-Biofilm Drugs" Microorganisms 8, no. 9: 1310. https://doi.org/10.3390/microorganisms8091310
APA StyleHall, D. C., Jr., Król, J. E., Cahill, J. P., Ji, H.-F., & Ehrlich, G. D. (2020). The Development of a Pipeline for the Identification and Validation of Small-Molecule RelA Inhibitors for Use as Anti-Biofilm Drugs. Microorganisms, 8(9), 1310. https://doi.org/10.3390/microorganisms8091310