Genome Sequence of Trichoderma lixii MUT3171, A Promising Strain for Mycoremediation of PAH-Contaminated Sites


 ,
, 
 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Microorganism and Growth Conditions
2.2. Laccases and Biosurfactants Analysis
2.3. DNA Extraction, Sequencing and Bioinformatics Analyses
3. Results and Discussion
3.1. Trichoderma lixii MUT3171 Genomic Parameters
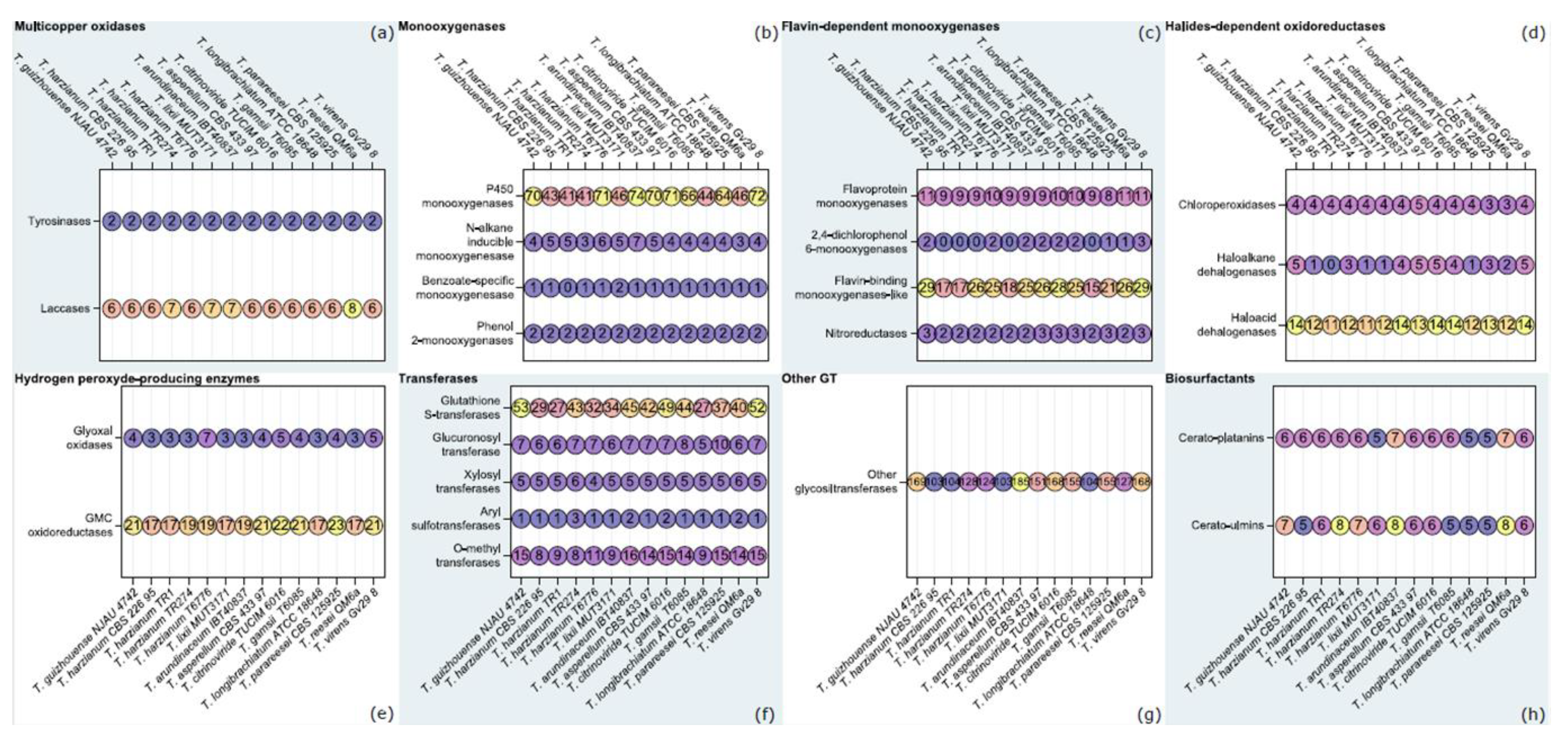
3.2. Orthology-Based Survey of T. lixii MUT3171 Degradative Enzymes
3.3. Unique Genetic Features of T. lixii MUT3171
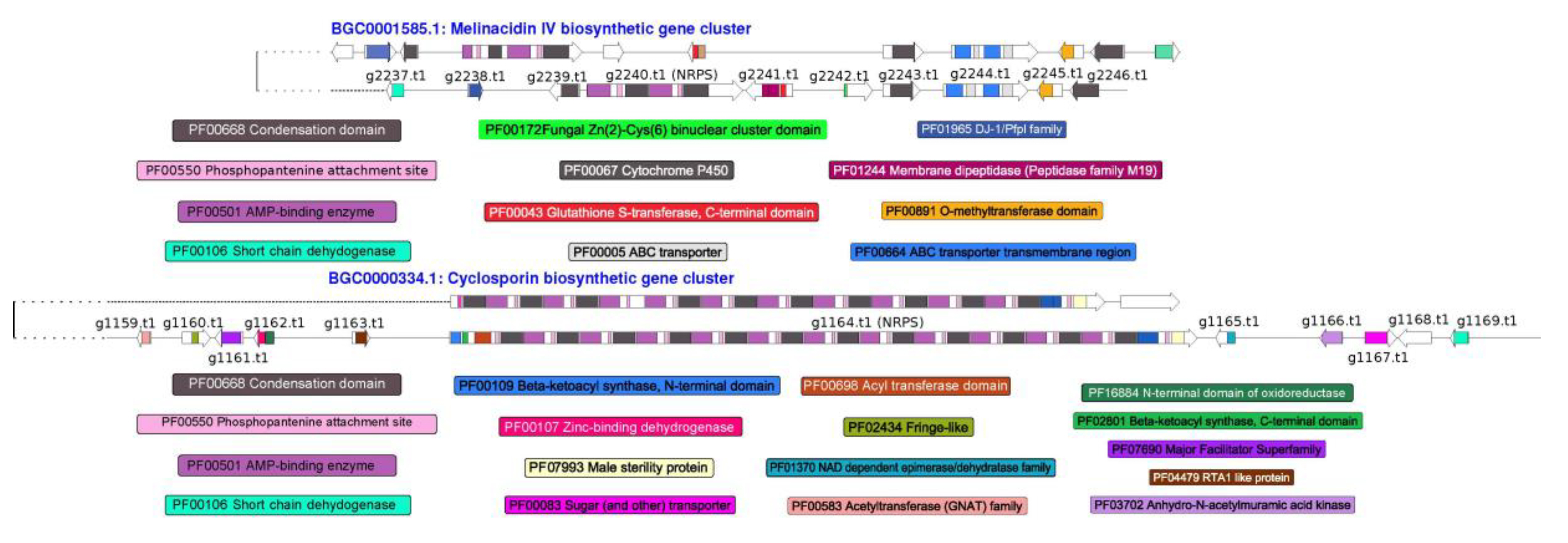
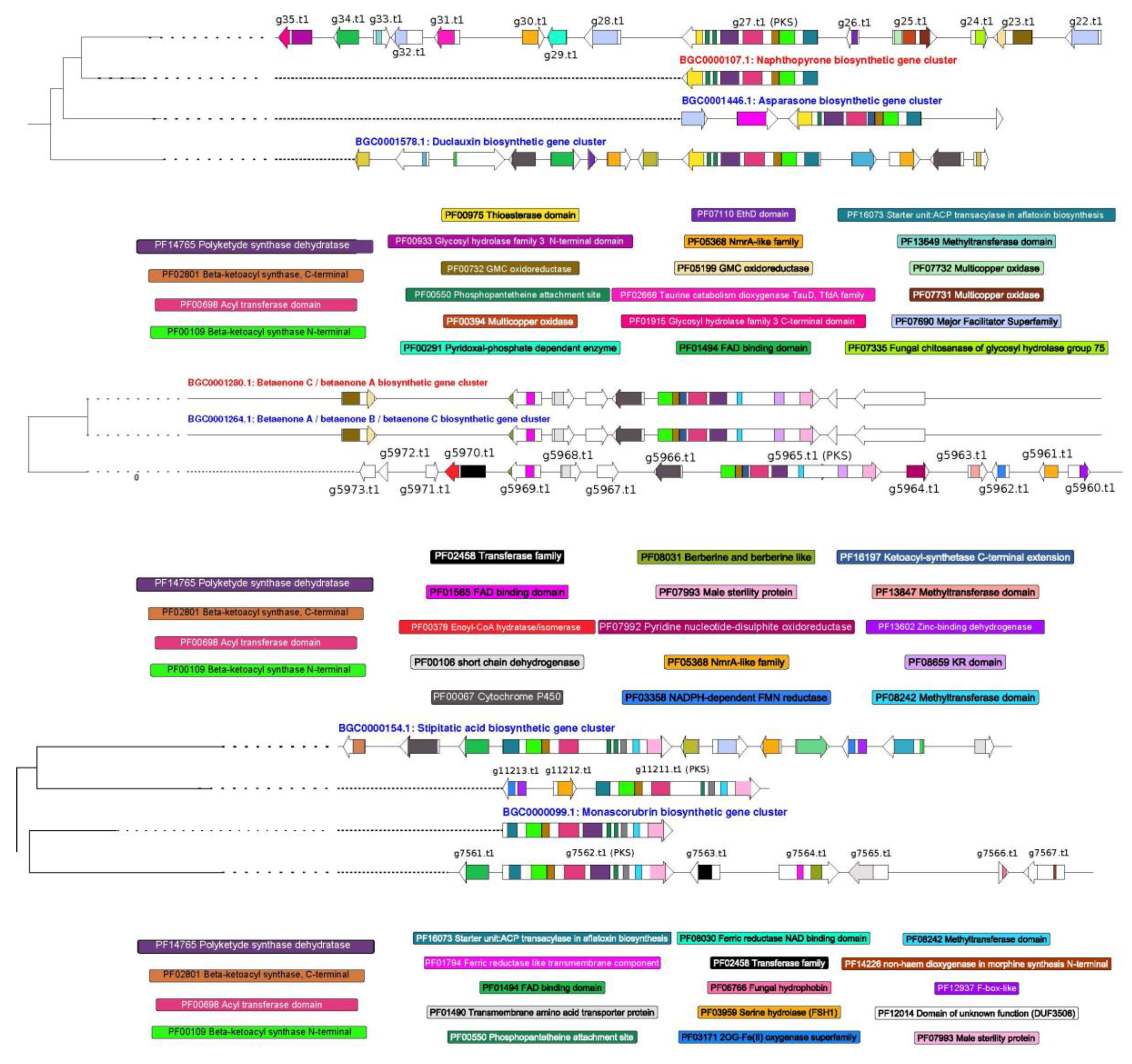
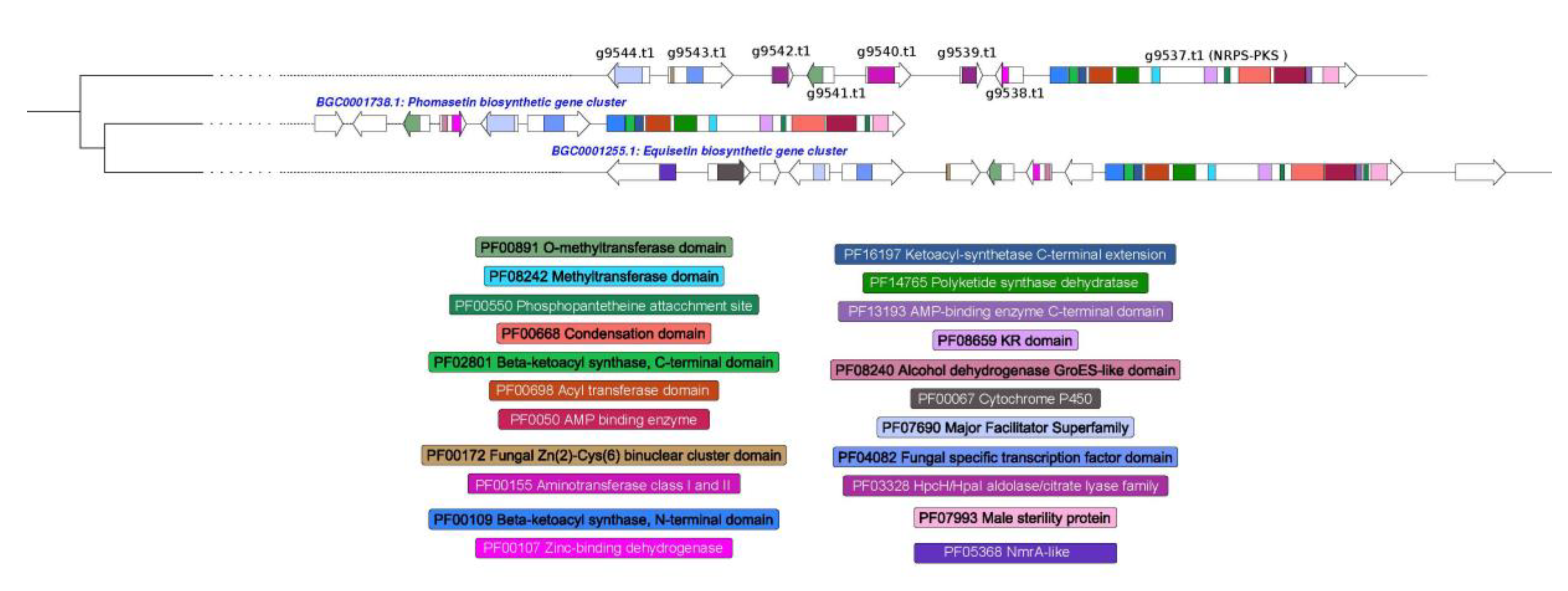
3.4. Prediction of Secondary Metabolism Genes in T. lixii MUT3171
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Saravanakumar, K.; Wang, M.-H. Isolation and molecular identification of Trichoderma species from wetland soil and their antagonistic activity against phytopathogens. Physiol. Mol. Plant Pathol. 2020, 109, 101458. [Google Scholar] [CrossRef]
- Katoch, M.; Singh, D.; Kapoor, K.K.; Vishwakarma, R.A. Trichoderma lixii (iiim-B4), an endophyte of Bacopa Monnieri L. producing peptaibols. BMC Microbiol. 2019, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Horta, M.A.C.; Filho, J.A.F.; Murad, N.F.; de Oliveira Santos, E.; dos Santos, C.A.; Mendes, J.S.; Brandão, M.M.; Azzoni, S.F.; de Souza, A.P. Network of proteins, enzymes and genes linked to biomass degradation shared by Trichoderma species. Sci. Rep. 2018, 8, 1341. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, C.P.; Steindorff, A.S.; Chenthamara, K.; Manganiello, G.; Henrissat, B.; Zhang, J.; Cai, F.; Kopchinskiy, A.G.; Kubicek, E.M.; Kuo, A.; et al. Evolution and comparative genomics of the most common Trichoderma species. BMC Genom. 2019, 20, 485. [Google Scholar] [CrossRef]
- Druzhinina, I.S.; Chenthamara, K.; Zhang, J.; Atanasova, L.; Yang, D.; Miao, Y.; Rahimi, M.J.; Grujic, M.; Cai, F.; Pourmehdi, S.; et al. Massive lateral transfer of genes encoding plant cell wall-degrading enzymes to the mycoparasitic fungus Trichoderma from its plant-associated hosts. PLoS Genet. 2018, 14, e1007322. [Google Scholar] [CrossRef]
- Zafra, G.; Cortés-Espinosa, D.V. Biodegradation of polycyclic aromatic hydrocarbons by Trichoderma species: A mini review. Environ. Sci. Pollut. Res. 2015, 22, 19426–19433. [Google Scholar] [CrossRef]
- Bosco, F.; Mollea, C. Mycoremediation in soil. Environ. Chem. Recent Pollut. Control. Approaches 2019. [Google Scholar] [CrossRef]
- Da Silva, A.C.S.; Dos Santos, P.N.; E Silva, T.A.L.; Andrade, R.F.S.; Campos-Takaki, G. Biosurfactant production by fungi as a sustainable alternative. Arq. Inst. Biol. 2018, 85. [Google Scholar] [CrossRef]
- Martinho, V.; dos Santos Lima, L.M.; Barros, C.A.; Ferrari, V.B.; Passarini, M.R.Z.; Santos, L.A.; de Souza Sebastianes, F.L.; Lacava, P.T.; de Vasconcellos, S.P. Enzymatic potential and biosurfactant production by endophytic fungi from mangrove forest in southeastern Brazil. AMB Express 2019, 9, 130. [Google Scholar] [CrossRef]
- Sena, H.H.; Sanches, M.A.; Rocha, D.F.S.; Segundo, W.O.P.F.; de Souza, É.S.; de Souza, J.V.B. Production of biosurfactants by soil fungi isolated from the Amazon forest. Int. J. Microbiol. 2018, 2018. [Google Scholar] [CrossRef]
- Barúa, J.E.; de la Cruz, M.; de Pedro, N.; Cautain, B.; Hermosa, R.; Cardoza, R.E.; Gutiérrez, S.; Monte, E.; Vicente, F.; Collado, I.G. Synthesis of trichodermin derivatives and their antimicrobial and cytotoxic activities. Molecules 2019, 24, 3811. [Google Scholar] [CrossRef] [PubMed]
- Sarsaiya, S.; Jain, A.; Kumar Awasthi, S.; Duan, Y.; Awasthi, M.K.; Shi, J. Microbial dynamics for lignocellulosic waste bioconversion and its importance with modern circular economy, challenges and future perspectives. Bioresour. Technol. 2019, 291, 121905. [Google Scholar] [CrossRef] [PubMed]
- Bodour, A.A.; Miller-Maier, R.M. Application of a modified drop-collapse technique for surfactant quantitation and screening of biosurfactant-producing microorganisms. J. Microbiol. Methods 1998, 32, 273–280. [Google Scholar] [CrossRef]
- Morikawa, M.; Hirata, Y.; Imanaka, T. A Study on the structure–function relationship of lipopeptide biosurfactants. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2000, 1488, 211–218. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects (accessed on 24 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 2013, 14, S7. [Google Scholar] [CrossRef]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucl. Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef]
- Beck, N.; Lang, B. MFannot, Organelle Genome Annotation Websever. 2010. Available online: http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl (accessed on 14 February 2020).
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucl. Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 30 January 2020).
- Smit, A.F.A.; Hubley, R. Repeatmodeler Open-1.0. Available online: http://www.repeatmasker.org/RepeatModeler/ (accessed on 30 January 2020).
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucl. Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 1–14. ISBN 978-1-4939-9173-0. [Google Scholar]
- Hoff, K.J.; Stanke, M. Predicting genes in single genomes with augustus. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Pellegrin, C.; Morin, E.; Martin, F.M.; Veneault-Fourrey, C. Comparative analysis of secretomes from ectomycorrhizal fungi with an emphasis on small-secreted proteins. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H. Predicting secretory proteins with SignalP. In Protein Function Prediction: Methods and Protocols; Kihara, D., Ed.; Springer: New York, NY, USA, 2017; pp. 59–73. ISBN 978-1-4939-7015-5. [Google Scholar]
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; Heijne, G.; von Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Gattiker, A.; Gasteiger, E.; Bairoch, A. ScanProsite: A reference implementation of a PROSITE scanning tool. Appl. Bioinform. 2002, 1, 107–108. [Google Scholar]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucl. Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile hmm searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucl. Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucl. Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucl. Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Harms, H.; Schlosser, D.; Wick, L.Y. Untapped potential: Exploiting fungi in bioremediation of hazardous chemicals. Nat. Rev. Microbiol. 2011, 9, 177–192. [Google Scholar] [CrossRef]
- Shanmugam, S.; Hari, A.; Ulaganathan, P.; Yang, F.; Krishnaswamy, S.; Wu, Y.-R. Potential of biohydrogen generation using the delignified lignocellulosic biomass by a newly identified thermostable laccase from Trichoderma asperellum strain Bplmbt1. Int. J. Hydrog. Energy 2018, 43, 3618–3628. [Google Scholar] [CrossRef]
- Cázares-García, S.V.; Vázquez-Garcidueñas, M.S.; Vázquez-Marrufo, G. Structural and phylogenetic analysis of laccases from Trichoderma: A bioinformatic approach. PLoS ONE 2013, 8, e55295. [Google Scholar] [CrossRef]
- Chakroun, H.; Mechichi, T.; Martinez, M.J.; Dhouib, A.; Sayadi, S. Purification and characterization of a novel laccase from the ascomycete Trichoderma atroviride: Application on bioremediation of phenolic compounds. Process. Biochem. 2010, 45, 507–513. [Google Scholar] [CrossRef]
- Sadhasivam, S.; Savitha, S.; Swaminathan, K.; Lin, F.-H. Production, purification and characterization of mid-redox potential laccase from a newly isolated Trichoderma harzianum Wl1. Process. Biochem. 2008, 43, 736–742. [Google Scholar] [CrossRef]
- Yada, G.M.; Shiraishi, I.S.; Dekker, R.F.H.; Schirmann, J.G.; Barbosa-Dekker, A.M.; de Araujo, I.C.; Abreu, L.M.; Daniel, J.F.S. Soil and entomopathogenic fungi with potential for biodegradation of insecticides: Degradation of flubendiamide in vivo by fungi and in vitro by laccase. Ann. Microbiol. 2019, 69, 1517–1529. [Google Scholar] [CrossRef]
- Nykiel-Szymańska, J.; Bernat, P.; Słaba, M. Potential of Trichoderma koningii to eliminate alachlor in the presence of copper ions. Ecotoxicol. Environ. Saf. 2018, 162, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, G.R.; Gopalakrishnan, T.; Sunkar, S. Production of laccase from Trichoderma harzianum and its application in dye decolourisation. Biocatal. Agric. Biotechnol. 2018, 16, 400–404. [Google Scholar] [CrossRef]
- Al Farraj, D.A.; Hadibarata, T.; Elshikh, M.S.; Al Khulaifi, M.M.; Kristanti, R.A. Biotransformation and degradation pathway of pyrene by filamentous soil fungus Trichoderma sp. F03. Water Air Soil Pollut. 2020, 231, 168. [Google Scholar] [CrossRef]
- Hadibarata, T.; Tachibana, S.; Itoh, K. Biodegradation of phenanthrene by fungi screened from nature. Pak. J. Biol. Sci. PJBS 2007, 10, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Daccò, C.; Girometta, C.; Asemoloye, M.D.; Carpani, G.; Picco, A.M.; Tosi, S. Key Fungal degradation patterns, enzymes and their applications for the removal of aliphatic hydrocarbons in polluted soils: A review. Int. Biodeterior. Biodegrad. 2020, 147, 104866. [Google Scholar] [CrossRef]
- Blesic, M.; Dichiarante, V.; Milani, R.; Linder, M.; Metrangolo, P. Evaluating the potential of natural surfactants in the petroleum industry: The case of hydrophobins. Pure Appl. Chem. 2018, 90, 305–314. [Google Scholar] [CrossRef]
- Pitocchi, R.; Cicatiello, P.; Birolo, L.; Piscitelli, A.; Bovio, E.; Varese, G.C.; Giardina, P. Cerato-platanins from marine fungi as effective protein biosurfactants and bioemulsifiers. Int. J. Mol. Sci. 2020, 21, 2913. [Google Scholar] [CrossRef]
- Sun, J.-Q.; Xu, L.; Liu, X.-Y.; Zhao, G.-F.; Cai, H.; Nie, Y.; Wu, X.-L. Functional genetic diversity and culturability of petroleum-degrading bacteria isolated from oil-contaminated soils. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.E.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes genomes: A test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Kohler, A.; Ohm, R.A.; Kuo, A.; Krützmann, J.; Morin, E.; Arend, M.; Barry, K.W.; Binder, M.; Choi, C.; et al. Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum Geophilum. Nat. Commun. 2016, 7, 12662. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Wang, H.; Liang, L.; Li, L.; Xiong, G.; Hu, Z. Phenanthrene degradation by the bacterium Pseudomonas stutzeri Jp1 under low oxygen condition. Int. Biodeterior. Biodegrad. 2017, 123, 121–126. [Google Scholar] [CrossRef]
- Presti, L.L.; Díaz, C.L.; Turrà, D.; Pietro, A.D.; Hampel, M.; Heimel, K.; Kahmann, R. A Conserved co-chaperone is required for virulence in fungal plant pathogens. New Phytol. 2016, 209, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Blasi, B.; Tafer, H.; Kustor, C.; Poyntner, C.; Lopandic, K.; Sterflinger, K. Genomic and transcriptomic analysis of the toluene degrading black yeast Cladophialophora immunda. Sci. Rep. 2017, 7, 11436. [Google Scholar] [CrossRef]
- Bakti, F.; Sasse, C.; Heinekamp, T.; Pócsi, I.; Braus, G.H. Heavy metal-induced Expression of PcaA provides cadmium tolerance to Aspergillus fumigatus and supports its virulence in the Galleria mellonella model. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Li, D.; Xu, X.; Hu, X.; Liu, Q.; Wang, Z.; Zhang, H.; Wang, H.; Wei, M.; Wang, H.; Liu, H.; et al. Genome-wide analysis and heavy metal-induced expression profiling of the HMA gene family in Populus trichocarpa. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Haas, H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. Mibig 2.0: A repository for biosynthetic gene clusters of known function. Nucl. Acids Res. 2020, 48, D454–D458. [Google Scholar] [CrossRef]
- Azam, A.; Anjum, T.; Irum, W. Trichoderma harzianum: A new fungal source for the production of cyclosporin A. Bangladesh J. Pharmacol. 2012, 7, 33–35. [Google Scholar] [CrossRef]
- Heine, D.; Holmes, N.A.; Worsley, S.F.; Santos, A.C.A.; Innocent, T.M.; Scherlach, K.; Patrick, E.H.; Yu, D.W.; Murrell, J.C.; Vieria, P.C.; et al. Chemical warfare between leafcutter ant symbionts and a co-evolved pathogen. Nat. Commun. 2018, 9, 2208. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Vinas, M.; Alsarrag, A.; Su, L.; Pfohl, K.; Rohlfs, M.; Schäfer, W.; Chen, W.; Karlovsky, P. Bis-naphthopyrone pigments protect filamentous ascomycetes from a wide range of predators. Nat. Commun. 2019, 10, 3579. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-X.; Tan, H.-B.; Liu, Y.; Chen, Y.-C.; Li, S.-N.; Sun, Z.-H.; Li, H.-H.; Qiu, S.-X.; Zhang, W.-M. Three new highly-oxygenated metabolites from the endophytic fungus Cytospora rhizophorae A761. Fitoterapia 2017, 117, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.; al Fahad, A.; Cai, M.; Song, Z.; Yehia, S.Y.; Lazarus, C.M.; Bailey, A.M.; Simpson, T.J.; Cox, R.J. Genetic, molecular, and biochemical basis of fungal tropolone biosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 7642–7647. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lam, C.-W.; Tam, E.W.T.; Lee, K.-C.; Yung, K.K.Y.; Leung, C.K.F.; Sze, K.-H.; Lau, S.K.P.; Yuen, K.-Y. The biosynthetic pathway for a thousand-year-old natural food colorant and citrinin in Penicillium marneffei. Sci. Rep. 2014, 4, 6728. [Google Scholar] [CrossRef]
- Reino, J.L.; Guerrero, R.F.; Hernández-Galán, R.; Collado, I.G. Secondary metabolites from species of the biocontrol agent Trichoderma. Phytochem. Rev. 2008, 7, 89–123. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Goetz, M.A.; Dombrowski, A.W.; Polishook, J.D.; Hazuda, D.J. Equisetin and a novel opposite stereochemical homolog phomasetin, two fungal metabolites as inhibitors of Hiv-1 integrase. Tetrahedron Lett. 1998, 39, 2243–2246. [Google Scholar] [CrossRef]
- Ahmed, E.; Holmström, S.J.M. Siderophores in environmental research: Roles and applications. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Estimated Value | Total Genome Coverage | ||
|---|---|---|---|---|
| Assembly size | ~40.89 Mbp | - | ||
| Number of scaffolds | 2142 | - | ||
| G+C content | ~49.4% | - | ||
| Genome gaps (Ns) | ~0.005% (2045 bp) | - | ||
| N50/L50 | 77,948 kbp/156 scaffolds | - | ||
| Number of genes | 11,923 | - | ||
| tRNAs | 182 | - | ||
| rRNAs | 8 s | 18 s | 28 s | - |
| 44 | 1 | 1 | - | |
| Small RNAs | 14 | - | ||
| Secreted proteins | 472 | - | ||
| Satellites | 14 | 0.02% | ||
| Simple repeats | 8820 | 0.89% | ||
| Low complexity regions | 1528 | 0.19% | ||
| Unclassified transposon sequences | 1499 | 0.71% | ||
| Retrotransposon sequences | SINEs | LINEs | LTR (Gypsy/DIRS1) | 0.83% |
| 15 | 226 | 536 | ||
| DNA transposon sequences | hobo-Activator | Tc1-IS630-Pogo | Others | 0.18% |
| 44 | 49 | 167 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venice, F.; Davolos, D.; Spina, F.; Poli, A.; Prigione, V.P.; Varese, G.C.; Ghignone, S. Genome Sequence of Trichoderma lixii MUT3171, A Promising Strain for Mycoremediation of PAH-Contaminated Sites. Microorganisms 2020, 8, 1258. https://doi.org/10.3390/microorganisms8091258
Venice F, Davolos D, Spina F, Poli A, Prigione VP, Varese GC, Ghignone S. Genome Sequence of Trichoderma lixii MUT3171, A Promising Strain for Mycoremediation of PAH-Contaminated Sites. Microorganisms. 2020; 8(9):1258. https://doi.org/10.3390/microorganisms8091258
Chicago/Turabian StyleVenice, Francesco, Domenico Davolos, Federica Spina, Anna Poli, Valeria Paola Prigione, Giovanna Cristina Varese, and Stefano Ghignone. 2020. "Genome Sequence of Trichoderma lixii MUT3171, A Promising Strain for Mycoremediation of PAH-Contaminated Sites" Microorganisms 8, no. 9: 1258. https://doi.org/10.3390/microorganisms8091258
APA StyleVenice, F., Davolos, D., Spina, F., Poli, A., Prigione, V. P., Varese, G. C., & Ghignone, S. (2020). Genome Sequence of Trichoderma lixii MUT3171, A Promising Strain for Mycoremediation of PAH-Contaminated Sites. Microorganisms, 8(9), 1258. https://doi.org/10.3390/microorganisms8091258