Heritability of Oral Microbiota and Immune Responses to Oral Bacteria

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Ethics Statement
2.3. Zygosity Determination
2.4. Saliva Collection and DNA Extraction for Microbiota Determination
2.5. Validation of DNA Extraction
2.6. Illumina Sequencing
2.7. Blood Collection and Serum Antibody Profile Determination
2.8. Statistics
2.8.1. Basic Descriptive Analysis
2.8.2. Generation of Standardized Abundance Variables and Definition of Detection
2.8.3. Intraclass Correlation Comparisons
2.8.4. Predicted Functions
2.8.5. Estimating Heritability
3. Results
3.1. Study Participants
3.2. Overall Sequencing Results
3.3. Species Retrieval by DNA Extraction Method
3.4. Overall Saliva Microbiota Diversity by Twin Zygosity
3.5. Intraclass Correlations of Saliva Microbiota Species in MZ versus DZ Twins
3.6. Heritability of Species Relative Abundance and Detection
3.7. Heritability of Predicted Functions
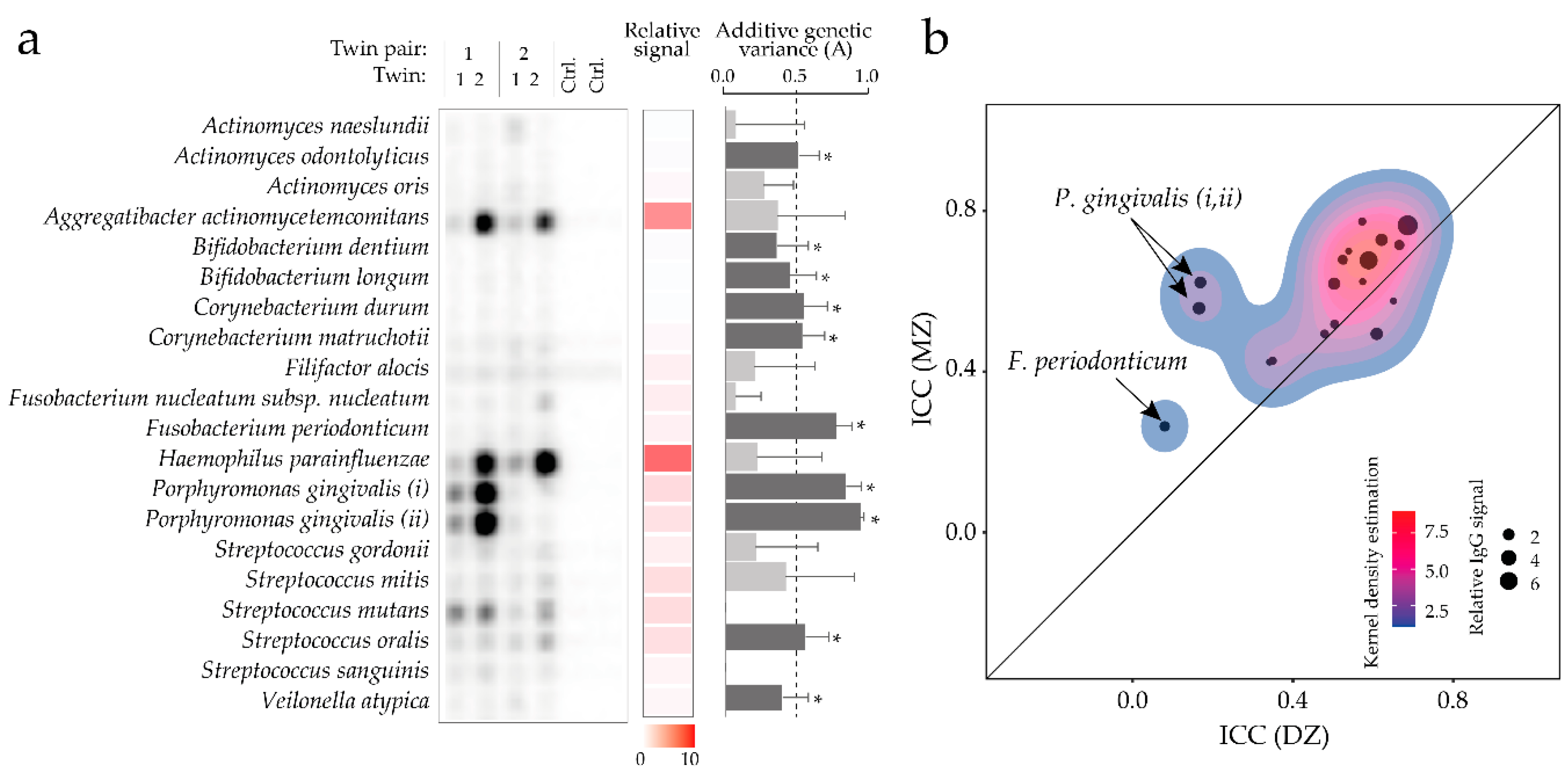
3.8. Heritability of Serum Antibody Levels against a Panel of Oral Bacteria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
Estimation of Multiple Testing Burden
References
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Garg, P.K.; Dubey, A.K. Insights into the human oral microbiome. Arch. Microbiol. 2018, 200, 525–540. [Google Scholar] [CrossRef]
- Nibali, L.; Bayliss-Chapman, J.; Almofareh, S.A.; Zhou, Y.; Divaris, K.; Vieira, A.R. What Is the Heritability of Periodontitis? A Systematic Review. J. Dent. Res. 2019, 98, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.R.; Feingold, E.; Wang, X.; Tcuenco, K.T.; Weeks, D.E.; DeSensi, R.S.; Polk, D.E.; Wendell, S.; Weyant, R.J.; Crout, R.; et al. Heritable patterns of tooth decay in the permanent dentition: Principal components and factor analyses. BMC Oral Health 2012, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Haworth, S.; Esberg, A.; Lif Holgerson, P.; Kuja-Halkola, R.; Timpson, N.J.; Magnusson, P.K.E.; Franks, P.W.; Johansson, I. Heritability of Caries Scores, Trajectories, and Disease Subtypes. J. Dent. Res. 2020, 99, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Bearfield, C.; Davenport, E.S.; Sivapathasundaram, V.; Allaker, R.P. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG 2002, 109, 527–533. [Google Scholar] [CrossRef]
- Dzidic, M.; Collado, M.C.; Abrahamsson, T.; Artacho, A.; Stensson, M.; Jenmalm, M.C.; Mira, A. Oral microbiome development during childhood: An ecological succession influenced by postnatal factors and associated with tooth decay. ISME J. 2018, 12, 2292–2306. [Google Scholar] [CrossRef]
- Kahharova, D.; Brandt, B.W.; Buijs, M.J.; Peters, M.; Jackson, R.; Eckert, G.; Katz, B.; Keels, M.A.; Levy, S.M.; Fontana, M.; et al. Maturation of the Oral Microbiome in Caries-Free Toddlers: A Longitudinal Study. J. Dent. Res. 2020, 99, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Siqueira, J.F., Jr.; Rocas, I.N. The Oral Microbiota in Health and Disease: An Overview of Molecular Findings. Methods Mol. Biol. 2017, 1537, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Corby, P.M.; Bretz, W.A.; Hart, T.C.; Filho, M.M.; Oliveira, B.; Vanyukov, M. Mutans streptococci in preschool twins. Arch. Oral Biol. 2005, 50, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Corby, P.M.; Bretz, W.A.; Hart, T.C.; Schork, N.J.; Wessel, J.; Lyons-Weiler, J.; Paster, B.J. Heritability of oral microbial species in caries-active and caries-free twins. Twin Res. Hum. Genet. 2007, 10, 821–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretz, W.A.; Corby, P.M.; Hart, T.C.; Costa, S.; Coelho, M.Q.; Weyant, R.J.; Robinson, M.; Schork, N.J. Dental caries and microbial acid production in twins. Caries Res. 2005, 39, 168–172. [Google Scholar] [CrossRef]
- Bretz, W.A.; Corby, P.M.; Melo, M.R.; Coelho, M.Q.; Costa, S.M.; Robinson, M.; Schork, N.J.; Drewnowski, A.; Hart, T.C. Heritability estimates for dental caries and sucrose sweetness preference. Arch. Oral Biol. 2006, 51, 1156–1160. [Google Scholar] [CrossRef]
- Du, Q.; Li, M.; Zhou, X.; Tian, K. A comprehensive profiling of supragingival bacterial composition in Chinese twin children and their mothers. Antonie Van Leeuwenhoek 2017, 110, 615–627. [Google Scholar] [CrossRef]
- Papapostolou, A.; Kroffke, B.; Tatakis, D.N.; Nagaraja, H.N.; Kumar, P.S. Contribution of host genotype to the composition of health-associated supragingival and subgingival microbiomes. J. Clin. Periodontol. 2011, 38, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.; Ribeiro, A.L.R.; Levine, A.P.; Pontikos, N.; Balloux, F.; Segal, A.W.; Roberts, A.P.; Smith, A.M. The Human Salivary Microbiome Is Shaped by Shared Environment Rather than Genetics: Evidence from a Large Family of Closely Related Individuals. MBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhang, M.; Li, J.; Li, Y.; Teng, F.; Jiang, H.; Du, M. Comparative Analysis of the Microbial Profiles in Supragingival Plaque Samples Obtained from Twins With Discordant Caries Phenotypes and Their Mothers. Front. Cell Infect. Microbiol. 2018, 8, 361. [Google Scholar] [CrossRef] [Green Version]
- Demmitt, B.A.; Corley, R.P.; Huibregtse, B.M.; Keller, M.C.; Hewitt, J.K.; McQueen, M.B.; Knight, R.; McDermott, I.; Krauter, K.S. Genetic influences on the human oral microbiome. BMC Genom. 2017, 18, 659. [Google Scholar] [CrossRef] [Green Version]
- Gomez, A.; Espinoza, J.L.; Harkins, D.M.; Leong, P.; Saffery, R.; Bockmann, M.; Torralba, M.; Kuelbs, C.; Kodukula, R.; Inman, J.; et al. Host Genetic Control of the Oral Microbiome in Health and Disease. Cell Host Microbe 2017, 22, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esberg, A.; Haworth, S.; Hasslof, P.; Lif Holgerson, P.; Johansson, I. Oral Microbiota Profile Associates with Sugar Intake and Taste Preference Genes. Nutrients 2020, 12, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagai, U.; Lichtenstein, P.; Pedersen, N.L.; Magnusson, P.K.E. The Swedish Twin Registry: Content and Management as a Research Infrastructure. Twin Res. Hum. Genet. 2019, 22, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anckarsater, H.; Lundstrom, S.; Kollberg, L.; Kerekes, N.; Palm, C.; Carlstrom, E.; Langstrom, N.; Magnusson, P.K.; Halldner, L.; Bolte, S.; et al. The Child and Adolescent Twin Study in Sweden (CATSS). Twin Res. Hum. Genet. 2011, 14, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Bokenberger, K.; Sjolander, A.; Dahl Aslan, A.K.; Karlsson, I.K.; Akerstedt, T.; Pedersen, N.L. Shift work and risk of incident dementia: A study of two population-based cohorts. Eur. J. Epidemiol. 2018, 33, 977–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannelius, U.; Gherman, L.; Makela, V.V.; Lindstedt, A.; Zucchelli, M.; Lagerberg, C.; Tybring, G.; Kere, J.; Lindgren, C.M. Large-scale zygosity testing using single nucleotide polymorphisms. Twin Res. Hum. Genet. 2007, 10, 604–625. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Chen, T.; Yu, W.H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, 2010, baq013. [Google Scholar] [CrossRef]
- Sakellari, D.; Socransky, S.S.; Dibart, S.; Eftimiadi, C.; Taubman, M.A. Estimation of serum antibody to subgingival species using checkerboard immunoblotting. Oral Microbiol. Immunol. 1997, 12, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Esberg, A.; Johansson, A.; Claesson, R.; Johansson, I. 43-Year Temporal Trends in Immune Response to Oral Bacteria in a Swedish Population. Pathogens 2020, 9, 544. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Beiko, R.G.; Langille, M.G.I. Predicting the Functional Potential of the Microbiome from Marker Genes Using PICRUSt. In Microbiome Analysis; Humana Press: New York, NY, USA, 2018; Volume 1849, pp. 169–177. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Falconer, D.S. Introduction to Quantitative Genetics; Long back; Oliver & Boyd: Edinburg, UK; London, UK, 1960; pp. 1–376. [Google Scholar]
- Kohler, H.P.; Behrman, J.R.; Schnittker, J. Social science methods for twins data: Integrating causality, endowments, and heritability. Biodemogr. Soc. Biol. 2011, 57, 88–141. [Google Scholar] [CrossRef] [Green Version]
- Neale, M.C.; Hunter, M.D.; Pritikin, J.N.; Zahery, M.; Brick, T.R.; Kirkpatrick, R.M.; Estabrook, R.; Bates, T.C.; Maes, H.H.; Boker, S.M. OpenMx 2.0: Extended Structural Equation and Statistical Modeling. Psychometrika 2016, 81, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Bonett, D.G.; Price, R.M. Inferential methods for the tetrachoric correlation coefficient. J. Educ. Behav. Stat. 2005, 30, 213–225. [Google Scholar] [CrossRef]
- Eriksson, L.; Lif Holgerson, P.; Johansson, I. Saliva and tooth biofilm bacterial microbiota in adolescents in a low caries community. Sci. Rep. 2017, 7, 5861. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, L.; Lif Holgerson, P.; Esberg, A.; Johansson, I. Microbial Complexes and Caries in 17-Year-Olds with and without Streptococcus mutans. J. Dent. Res. 2018, 97, 275–282. [Google Scholar] [CrossRef]
- Bockmann, M.R.; Harris, A.V.; Bennett, C.N.; Odeh, R.; Hughes, T.E.; Townsend, G.C. Timing of colonization of caries-producing bacteria: An approach based on studying monozygotic twin pairs. Int. J. Dent. 2011, 2011, 571573. [Google Scholar] [CrossRef] [Green Version]
- Ooi, G.; Townsend, G.; Seow, W.K. Bacterial colonization, enamel defects and dental caries in 4–6-year-old mono- and dizygotic twins. Int. J. Paediatr. Dent. 2014, 24, 152–160. [Google Scholar] [CrossRef]
- Si, J.; Lee, C.; Ko, G. Oral Microbiota: Microbial Biomarkers of Metabolic Syndrome Independent of Host Genetic Factors. Front. Cell Infect. Microbiol. 2017, 7, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Esberg, A.; Sheng, N.; Marell, L.; Claesson, R.; Persson, K.; Boren, T.; Stromberg, N. Streptococcus Mutans Adhesin Biotypes that Match and Predict Individual Caries Development. EBioMedicine 2017, 24, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskitalo, K.; Tuorila, H.; Spector, T.D.; Cherkas, L.F.; Knaapila, A.; Silventoinen, K.; Perola, M. Same genetic components underlie different measures of sweet taste preference. Am. J. Clin. Nutr. 2007, 86, 1663–1669. [Google Scholar] [CrossRef]
- Pallister, T.; Sharafi, M.; Lachance, G.; Pirastu, N.; Mohney, R.P.; MacGregor, A.; Feskens, E.J.; Duffy, V.; Spector, T.D.; Menni, C. Food Preference Patterns in a UK Twin Cohort. Twin Res. Hum. Genet. 2015, 18, 793–805. [Google Scholar] [CrossRef] [Green Version]
- Treur, J.L.; Boomsma, D.I.; Ligthart, L.; Willemsen, G.; Vink, J.M. Heritability of high sugar consumption through drinks and the genetic correlation with substance use. Am. J. Clin. Nutr. 2016, 104, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aruni, A.W.; Mishra, A.; Dou, Y.; Chioma, O.; Hamilton, B.N.; Fletcher, H.M. Filifactor alocis-a new emerging periodontal pathogen. Microbes Infect. 2015, 17, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Ayala Herrera, J.L.; Apreza Patron, L.; Martinez Martinez, R.E.; Dominguez Perez, R.A.; Abud Mendoza, C.; Hernandez Castro, B. Filifactor alocis and Dialister pneumosintes in a Mexican population affected by periodontitis and rheumatoid arthritis: An exploratory study. Microbiol. Immunol. 2019, 63, 392–395. [Google Scholar] [CrossRef]
- Engstrom, M.; Eriksson, K.; Lee, L.; Hermansson, M.; Johansson, A.; Nicholas, A.P.; Gerasimcik, N.; Lundberg, K.; Klareskog, L.; Catrina, A.I.; et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J. Transl. Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Gomez-Banuelos, E.; Mukherjee, A.; Darrah, E.; Andrade, F. Rheumatoid Arthritis-Associated Mechanisms of Porphyromonas gingivalis and Aggregatibacter Actinomycetemcomitans. J. Clin. Med. 2019, 8, 1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Atienza, E.; Flak, M.B.; Sirr, J.; Paramonov, N.A.; Aduse-Opoku, J.; Pitzalis, C.; Curtis, M.A. The, P. gingivalis Autocitrullinome Is Not a Target for ACPA in Early Rheumatoid Arthritis. J. Dent. Res. 2020, 99, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, S.C.; Kesavalu, L.; Walker, S.; Genco, C.A. Virulence factors of Porphyromonas gingivalis. Periodontol. 2000 1999, 20, 168–238. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas gingivalis: Major periodontopathic pathogen overview. J. Immunol Res. 2014, 2014, 476068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, M.; Ohara, N. Molecular mechanisms of Porphyromonas gingivalis-host cell interaction on periodontal diseases. Jpn. Dent. Sci. Rev. 2017, 53, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissbrod, O.; Rothschild, D.; Barkan, E.; Segal, E. Host genetics and microbiome associations through the lens of genome wide association studies. Curr. Opin. Microbiol. 2018, 44, 9–19. [Google Scholar] [CrossRef]
- Konig, M.F. The microbiome in autoimmune rheumatic disease. Best Pract. Res. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Fak, F.; Tremaroli, V.; Bergstrom, G.; Backhed, F. Oral microbiota in patients with atherosclerosis. Atherosclerosis 2015, 243, 573–578. [Google Scholar] [CrossRef]
- Minty, M.; Canceil, T.; Serino, M.; Burcelin, R.; Terce, F.; Blasco-Baque, V. Oral microbiota-induced periodontitis: A new risk factor of metabolic diseases. Rev. Endocr. Metab. Disord. 2019, 20, 449–459. [Google Scholar] [CrossRef]
- Humphrey, S.P.; Williamson, R.T. A review of saliva: Normal composition, flow, and function. J. Prosthet. Dent. 2001, 85, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Esberg, A.; Haworth, S.; Holgerson, P.L.; Johansson, I. Allelic Variation in Taste Genes Is Associated with Taste and Diet Preferences and Dental Caries. Nutrients 2019, 11, 1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, I.; Witkowska, E.; Kaveh, B.; Lif Holgerson, P.; Tanner, A.C. The Microbiome in Populations with a Low and High Prevalence of Caries. J. Dent. Res. 2016, 95, 80–86. [Google Scholar] [CrossRef]
- Donlin, L.T.; Park, S.H.; Giannopoulou, E.; Ivovic, A.; Park-Min, K.H.; Siegel, R.M.; Ivashkiv, L.B. Insights into rheumatic diseases from next-generation sequencing. Nat. Rev. Rheumatol. 2019, 15, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Belstrom, D. The salivary microbiota in health and disease. J. Oral Microbiol. 2020, 12, 1723975. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | Microbiota Characterization | Antibody Screening | ||||
|---|---|---|---|---|---|---|
| MZ | DZ-SS | DZ-OS | MZ | DZ-SS | DZ-OS | |
| Number of twin pairs | 71 | 68 | 70 | 70 | 69 | 70 |
| Birth year interval | 1993–2001 | 1993–2001 | 1993–2001 | 1923–1958 | 1917–1957 | 1926–1958 |
| Sampling year interval | 2006–2017 | 2007–2010 | 2006–2011 | 2005–2006 | 2004–2008 | 2004–2008 |
| Storage year, mean (95% CI) | 10.5 (10.2, 10.7) | 10.6 (10.4, 10.7) | 10.5 (10.3, 10.7) | 13.4 (13.3, 13.6) | 14.0 (13.8, 14.2) | 13.9 (13.7, 14.1) |
| Sex, % females | 49.3 | 58.8 | 50.0 | 65.7 | 62.3 | 50.0 |
| Screening age, mean (95% CI) | 12.9 (12.5, 13.2) | 12.1 (11.7, 12.4) | 12.1 (11.7, 12.4) | 62.7 (61.4, 64.0) | 65.4 (64.0, 66.8) | 63.9 (62.5, 65.2) |
| DMFS, mean (95% CI) a | 1.3 (0.6, 2.1) | 1.9 (1.2, 2.7) | 3.0 (2.2., 3.7) | 86.8 (82.4, 91.1) | 88.8 (84.5, 93.1) | 86.9 (82.6, 91.2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esberg, A.; Haworth, S.; Kuja-Halkola, R.; Magnusson, P.K.E.; Johansson, I. Heritability of Oral Microbiota and Immune Responses to Oral Bacteria. Microorganisms 2020, 8, 1126. https://doi.org/10.3390/microorganisms8081126
Esberg A, Haworth S, Kuja-Halkola R, Magnusson PKE, Johansson I. Heritability of Oral Microbiota and Immune Responses to Oral Bacteria. Microorganisms. 2020; 8(8):1126. https://doi.org/10.3390/microorganisms8081126
Chicago/Turabian StyleEsberg, Anders, Simon Haworth, Ralf Kuja-Halkola, Patrik K.E. Magnusson, and Ingegerd Johansson. 2020. "Heritability of Oral Microbiota and Immune Responses to Oral Bacteria" Microorganisms 8, no. 8: 1126. https://doi.org/10.3390/microorganisms8081126
APA StyleEsberg, A., Haworth, S., Kuja-Halkola, R., Magnusson, P. K. E., & Johansson, I. (2020). Heritability of Oral Microbiota and Immune Responses to Oral Bacteria. Microorganisms, 8(8), 1126. https://doi.org/10.3390/microorganisms8081126