Helicobacter pylori Colonization Drives Urokinase Receptor (uPAR) Expression in Murine Gastric Epithelium During Early Pathogenesis

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Ethics Statement
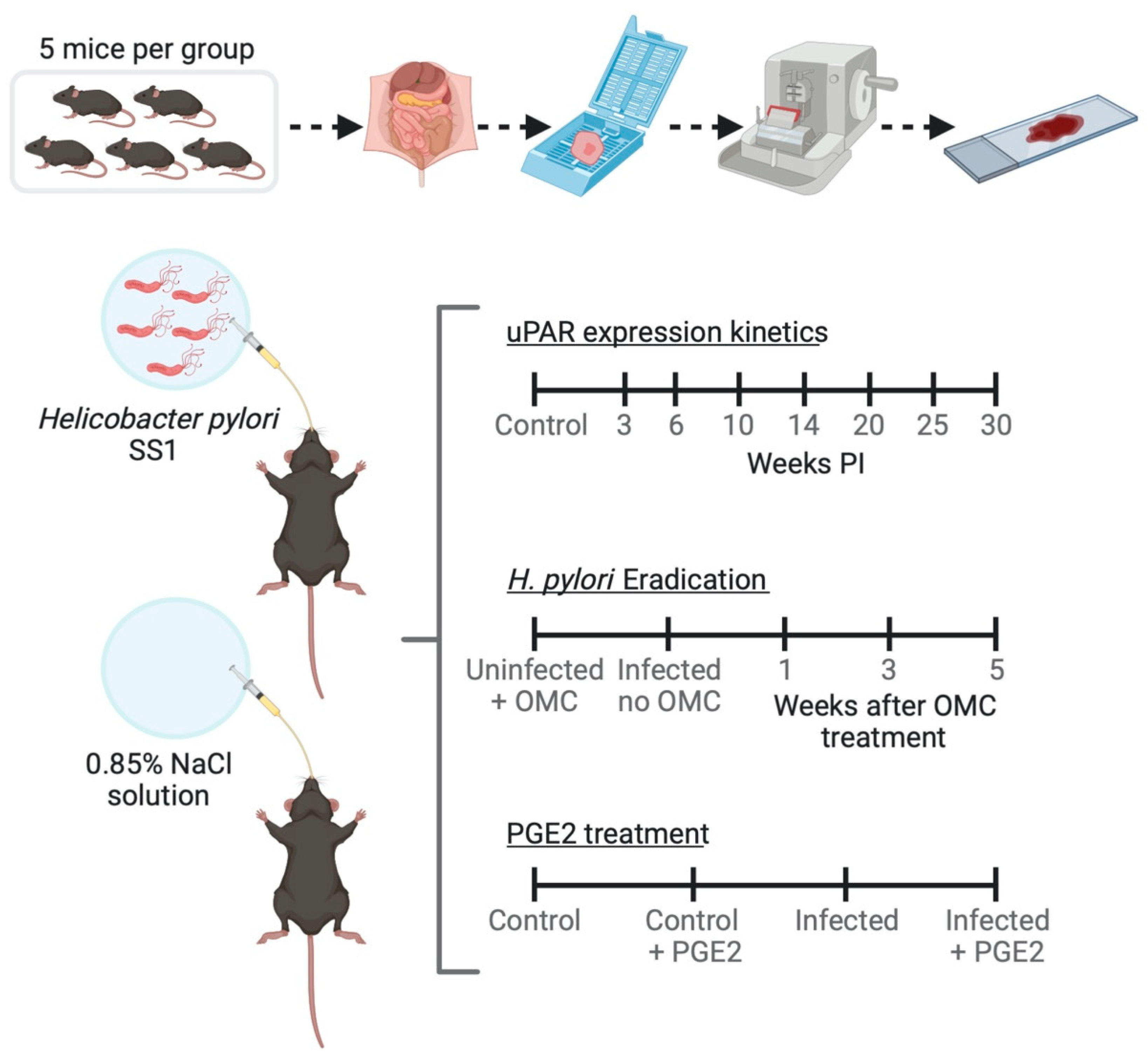
2.2. Mice
2.3. H. pylori Culture and Experimental Infections
2.4. Antimicrobial Therapy for H. pylori Eradication in Mice
2.5. Pharmacological Treatment of Mice with Prostaglandin E2 (PGE2) Analogs
2.6. Resection and Processing of Gastric Tissue for Histology
2.7. Immunohistochemical Detection of uPAR, Ki67, and Inflammation Markers in Gastric Mucosa
2.8. In Vitro Transcription and In Situ Hybridization for uPA
2.9. Co-Culture Conditions of Human Gastric Cancer Cell Lines and H. pylori Strains In Vitro
2.10. H. pylori Colonization, Histopathology and Immunohistochemistry Evaluations
2.11. Statistical Analysis
3. Results
3.1. Histopathological Changes Induced by H. pylori Infection
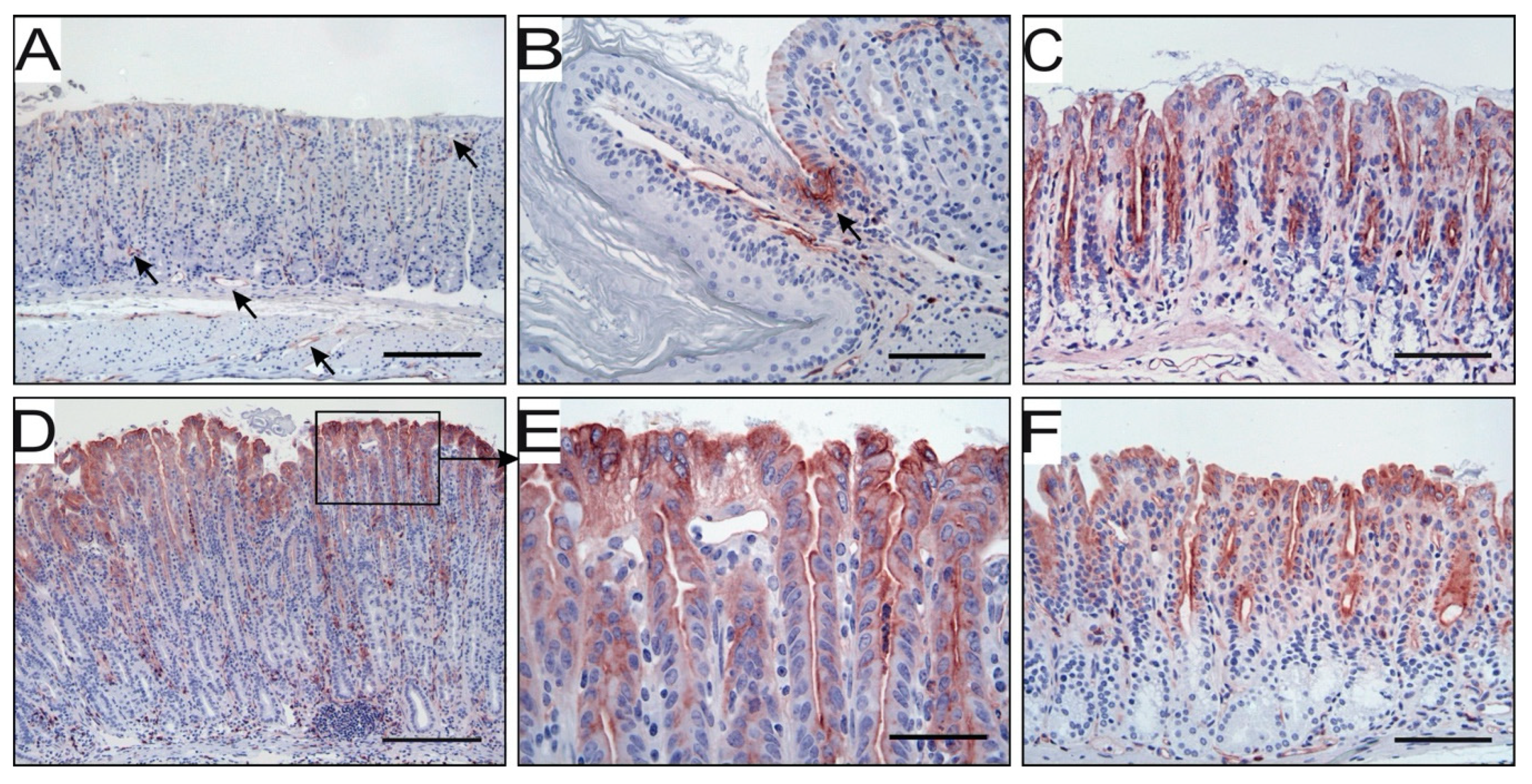
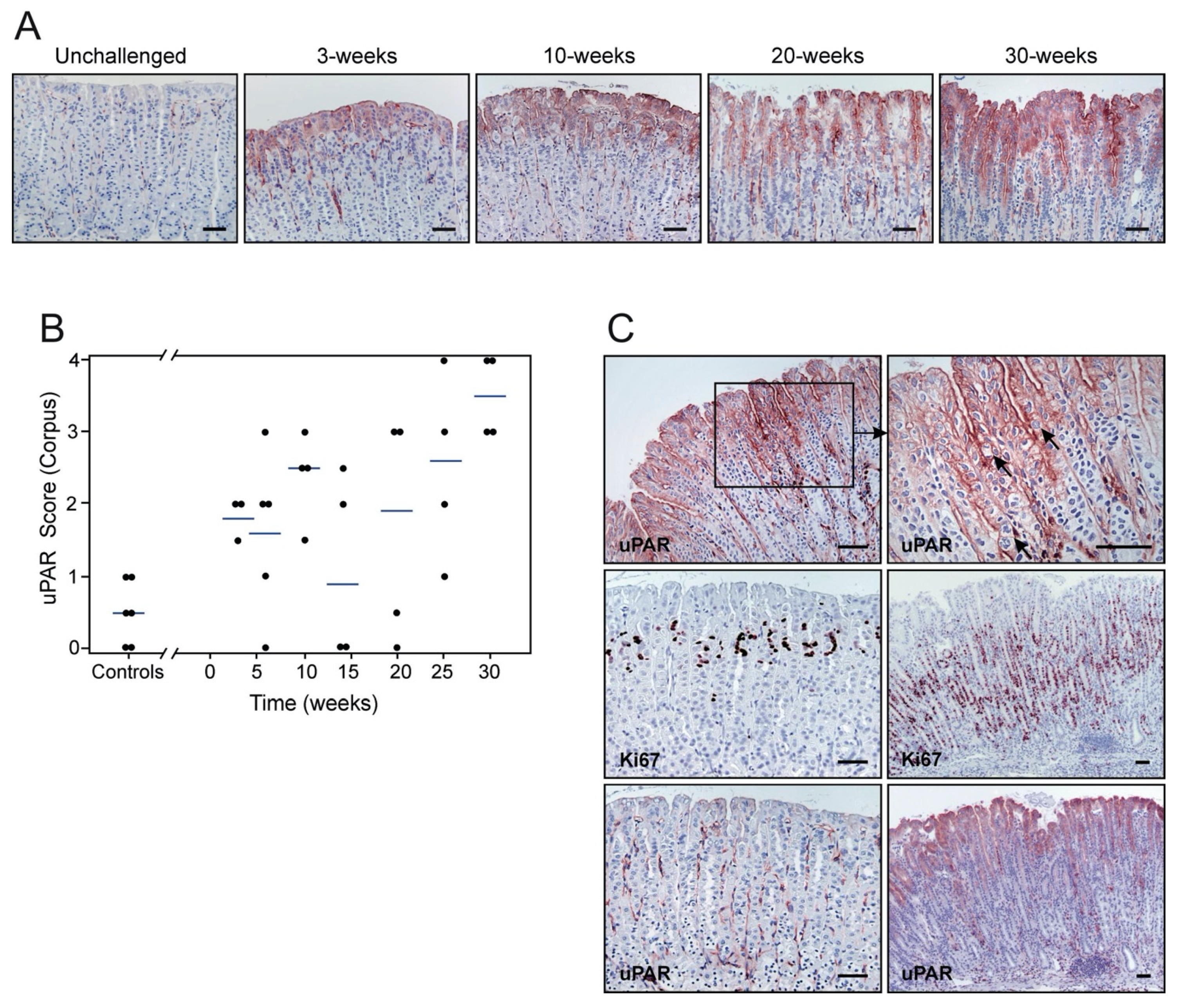
3.2. H. pylori Infection Induces uPAR Expression in Gastric Epithelial Cells
3.3. uPAR Expression Levels, Inflammation, and Cell Proliferation Increase with the Duration of H. pylori Infection
3.4. uPA is Not UpRegulated in Gastric Mucosa Upon H. pylori Infection
3.5. Antimicrobial Eradication of H. pylori Infection Downregulates uPAR Expression
3.6. Treatment with PGE2 Attenuates Gastric Immunopathology but Does Not Abrogate uPAR Expression in H. pylori-infected Mice
3.7. H. pylori Induces uPAR Expression in Co-Cultured Gastric Adenocarcinoma Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef]
- O’Connor, A.; O’Morain, C.A.; Ford, A.C. Population screening and treatment of Helicobacter pylori infection. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Kyburz, A.; Müller, A. Helicobacter pylori and Extragastric Diseases. Curr. Top. Microbiol. Immunol. 2017, 400, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Taye, B.; Enquselassie, F.; Tsegaye, A.; Amberbir, A.; Medhin, G.; Fogarty, A.; Robinson, K.; Davey, G. Association between infection with Helicobacter pylori and atopy in young Ethiopian children: A longitudinal study. Clin. Exp. Allergy 2017, 47, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the human stomach: Persistence strategies of the bacterial pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.T.; Crabtree, J.E. Immunology of Helicobacter pylori: Insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007, 133, 288–308. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L. Helicobacter pylori Diversity and Gastric Cancer Risk. MBio 2016, 7, e01869-15. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef]
- Kriegbaum, M.C.; Persson, M.; Haldager, L.; Alpízar-Alpízar, W.; Jacobsen, B.; Gårdsvoll, H.; Kjær, A.; Ploug, M. Rational targeting of the urokinase receptor (uPAR): Development of antagonists and non-invasive imaging probes. Curr. Drug Targets 2011, 12, 1711–1728. [Google Scholar] [CrossRef]
- Alpizar-Alpizar, W.; Christensen, I.J.; Santoni-Rugiu, E.; Skarstein, A.; Ovrebo, K.; Illemann, M.; Laerum, O.D. Urokinase plasminogen activator receptor on invasive cancer cells: A prognostic factor in distal gastric adenocarcinoma. Int. J. Cancer 2012, 131, E329–E336. [Google Scholar] [CrossRef] [PubMed]
- Brungs, D.; Chen, J.; Aghmesheh, M.; Vine, K.L.; Becker, T.M.; Carolan, M.G.; Ranson, M. The urokinase plasminogen activation system in gastroesophageal cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 23099–23109. [Google Scholar] [CrossRef] [PubMed]
- Lund, I.K.; Illemann, M.; Thurison, T.; Christensen, I.J.; Høyer-Hansen, G. uPAR as anti-cancer target: Evaluation of biomarker potential, histological localization, and antibody-based therapy. Curr. Drug Targets 2011, 12, 1744–1760. [Google Scholar] [CrossRef] [PubMed]
- Gårdsvoll, H.; Kjaergaard, M.; Jacobsen, B.; Kriegbaum, M.C.; Huang, M.; Ploug, M. Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor. J. Biol. Chem. 2011, 286, 43515–43526. [Google Scholar] [CrossRef]
- Madsen, C.D.; Ferraris, G.M.; Andolfo, A.; Cunningham, O.; Sidenius, N. uPAR-induced cell adhesion and migration: Vitronectin provides the key. J. Cell Biol. 2007, 177, 927–939. [Google Scholar] [CrossRef]
- Chaurasia, P.; Aguirre-Ghiso, J.A.; Liang, O.D.; Gardsvoll, H.; Ploug, M.; Ossowski, L. A region in urokinase plasminogen receptor domain III controlling a functional association with alpha5beta1 integrin and tumor growth. J. Biol. Chem. 2006, 281, 14852–14863. [Google Scholar] [CrossRef]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Persson, M.; Madsen, J.; Østergaard, S.; Jensen, M.M.; Jørgensen, J.T.; Juhl, K.; Lehmann, C.; Ploug, M.; Kjaer, A. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: Implications for visualizing cancer invasion. J. Nucl. Med. 2012, 53, 138–145. [Google Scholar] [CrossRef]
- Persson, M.; Skovgaard, D.; Brandt-Larsen, M.; Christensen, C.; Madsen, J.; Nielsen, C.H.; Thurison, T.; Klausen, T.L.; Holm, S.; Loft, A.; et al. First-in-human uPAR PET: Imaging of Cancer Aggressiveness. Theranostics 2015, 5, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M. Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist. Theranostics 2013, 3, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- El-Etr, S.H.; Mueller, A.; Tompkins, L.S.; Falkow, S.; Merrell, D.S. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J. Infect. Dis. 2004, 190, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, K.; Salama, N.R.; Tompkins, L.S.; Falkow, S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15136–15141. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sierra, R.A.; McGee, D.J.; Zabaleta, J. Transcriptional profiling of gastric epithelial cells infected with wild type or arginase-deficient Helicobacter pylori. BMC Microbiol. 2012, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, A.R.; Tao, H.; Carloni, E.; Sepulveda, J.; Graham, D.Y.; Peterson, L.E. Screening of gene expression profiles in gastric epithelial cells induced by Helicobacter pylori using microarray analysis. Aliment. Pharmacol. Ther. 2002, 16 (Suppl. S2), 145–157. [Google Scholar] [CrossRef]
- Kenny, S.; Duval, C.; Sammut, S.J.; Steele, I.; Pritchard, D.M.; Atherton, J.C.; Argent, R.H.; Dimaline, R.; Dockray, G.J.; Varro, A. Increased expression of the urokinase plasminogen activator system by Helicobacter pylori in gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G431–G441. [Google Scholar] [CrossRef][Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Alpízar-Alpízar, W.; Nielsen, B.S.; Sierra, R.; Illemann, M.; Ramírez, J.A.; Arias, A.; Durán, S.; Skarstein, A.; Ovrebo, K.; Lund, L.R.; et al. Urokinase plasminogen activator receptor is expressed in invasive cells in gastric carcinomas from high- and low-risk countries. Int. J. Cancer 2010, 126, 405–415. [Google Scholar] [CrossRef]
- Gyetko, M.R.; Sud, S.; Kendall, T.; Fuller, J.A.; Newstead, M.W.; Standiford, T.J. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J. Immunol. 2000, 165, 1513–1519. [Google Scholar] [CrossRef]
- Hovius, J.W.; Bijlsma, M.F.; van der Windt, G.J.; Wiersinga, W.J.; Boukens, B.J.; Coumou, J.; Oei, A.; de Beer, R.; de Vos, A.F.; van’t Veer, C.; et al. The urokinase receptor (uPAR) facilitates clearance of Borrelia burgdorferi. PLoS Pathog. 2009, 5, e1000447. [Google Scholar] [CrossRef] [PubMed]
- Rijneveld, A.W.; Levi, M.; Florquin, S.; Speelman, P.; Carmeliet, P.; van Der Poll, T. Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. J. Immunol. 2002, 168, 3507–3511. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Kager, L.M.; Hovius, J.W.; van der Windt, G.J.; de Vos, A.F.; Meijers, J.C.; Roelofs, J.J.; Dondorp, A.; Levi, M.; Day, N.P.; et al. Urokinase receptor is necessary for bacterial defense against pneumonia-derived septic melioidosis by facilitating phagocytosis. J. Immunol. 2010, 184, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Rickman, B.; Rogers, A.B.; Muthupalani, S.; Takaishi, S.; Yang, P.; Wang, T.C.; Fox, J.G. Combination of sulindac and antimicrobial eradication of Helicobacter pylori prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2009, 69, 8166–8174. [Google Scholar] [CrossRef]
- van Zanten, S.J.; Kolesnikow, T.; Leung, V.; O’Rourke, J.L.; Lee, A. Gastric transitional zones, areas where Helicobacter treatment fails: Results of a treatment trial using the Sydney strain mouse model. Antimicrob. Agents Chemother. 2003, 47, 2249–2255. [Google Scholar] [CrossRef]
- Toller, I.M.; Hitzler, I.; Sayi, A.; Mueller, A. Prostaglandin E2 prevents Helicobacter-induced gastric preneoplasia and facilitates persistent infection in a mouse model. Gastroenterology 2010, 138, 1455–1467, 1467.e1451–e1454. [Google Scholar] [CrossRef]
- Solberg, H.; Ploug, M.; Høyer-Hansen, G.; Nielsen, B.S.; Lund, L.R. The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling. J. Histochem. Cytochem. 2001, 49, 237–246. [Google Scholar] [CrossRef]
- Kristensen, P.; Eriksen, J.; Danø, K. Localization of urokinase-type plasminogen activator messenger RNA in the normal mouse by in situ hybridization. J. Histochem. Cytochem. 1991, 39, 341–349. [Google Scholar] [CrossRef]
- Illemann, M.; Bird, N.; Majeed, A.; Laerum, O.D.; Lund, L.R.; Danø, K.; Nielsen, B.S. Two distinct expression patterns of urokinase, urokinase receptor and plasminogen activator inhibitor-1 in colon cancer liver metastases. Int. J. Cancer 2009, 124, 1860–1870. [Google Scholar] [CrossRef]
- Watson, S.A.; Durrant, L.G.; Wencyk, P.M.; Watson, A.L.; Morris, D.L. Intracellular gastrin in human gastrointestinal tumor cells. J. Natl. Cancer Inst. 1991, 83, 866–871. [Google Scholar] [CrossRef]
- Gårdsvoll, H.; Werner, F.; Søndergaard, L.; Danø, K.; Ploug, M. Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Schneider 2 cells after deletion of glycosylation-sites. Protein Expr. Purif. 2004, 34, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Goldenring, J.R.; Dangler, C.; Ito, S.; Mueller, A.; Jeon, W.K.; Koh, T.J.; Fox, J.G. Mice lacking secretory phospholipase A2 show altered apoptosis and differentiation with Helicobacter felis infection. Gastroenterology 1998, 114, 675–689. [Google Scholar] [CrossRef]
- Rogers, A.B. Histologic scoring of gastritis and gastric cancer in mouse models. Methods Mol. Biol. 2012, 921, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, J.; Mizokami, Y.; Takahashi, K.; Nakajima, K.; Ohtsubo, T.; Miura, S.; Narasaka, T.; Takeyama, H.; Omata, T.; Shimokobe, K.; et al. Expressions of urokinase-type plasminogen activator, its receptor and plasminogen activator inhibitor-1 in gastric cancer cells and effects of Helicobacter pylori. Scand. J. Gastroenterol. 2005, 40, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Yoo, H.S.; Kim, M.Y.; Jang, H.J.; Baek, M.K.; Kim, H.R.; Kim, K.K.; Shin, B.A.; Ahn, B.W.; Jung, Y.D. Helicobacter pylori stimulates urokinase plasminogen activator receptor expression and cell invasiveness through reactive oxygen species and NF-kappaB signaling in human gastric carcinoma cells. Int. J. Mol. Med. 2007, 19, 689–697. [Google Scholar]
- Kim, M.H.; Yoo, H.S.; Chang, H.J.; Hong, M.H.; Kim, H.D.; Chung, I.J.; Shin, B.A.; Cho, M.J.; Ahn, B.W.; Jung, Y.D. Urokinase plasminogen activator receptor is upregulated by Helicobacter pylori in human gastric cancer AGS cells via ERK, JNK, and AP-1. Biochem. Biophys. Res. Commun. 2005, 333, 874–880. [Google Scholar] [CrossRef]
- Connolly, B.M.; Choi, E.Y.; Gårdsvoll, H.; Bey, A.L.; Currie, B.M.; Chavakis, T.; Liu, S.; Molinolo, A.; Ploug, M.; Leppla, S.H.; et al. Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation. Blood 2010, 116, 1593–1603. [Google Scholar] [CrossRef]
- Danø, K.; Behrendt, N.; Høyer-Hansen, G.; Johnsen, M.; Lund, L.R.; Ploug, M.; Rømer, J. Plasminogen activation and cancer. Thromb. Haemost. 2005, 93, 676–681. [Google Scholar] [CrossRef]
- Heiss, M.M.; Allgayer, H.; Gruetzner, K.U.; Funke, I.; Babic, R.; Jauch, K.W.; Schildberg, F.W. Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: A reference to early systemic disease in solid cancer. Nat. Med. 1995, 1, 1035–1039. [Google Scholar] [CrossRef]
- Croxen, M.A.; Sisson, G.; Melano, R.; Hoffman, P.S. The Helicobacter pylori chemotaxis receptor TlpB (HP0103) is required for pH taxis and for colonization of the gastric mucosa. J. Bacteriol. 2006, 188, 2656–2665. [Google Scholar] [CrossRef]
- Schreiber, S.; Konradt, M.; Groll, C.; Scheid, P.; Hanauer, G.; Werling, H.O.; Josenhans, C.; Suerbaum, S. The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. USA 2004, 101, 5024–5029. [Google Scholar] [CrossRef] [PubMed]
- Falk, P.; Roth, K.A.; Borén, T.; Westblom, T.U.; Gordon, J.I.; Normark, S. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc. Natl. Acad. Sci. USA 1993, 90, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Hessey, S.J.; Spencer, J.; Wyatt, J.I.; Sobala, G.; Rathbone, B.J.; Axon, A.T.; Dixon, M.F. Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut 1990, 31, 134–138. [Google Scholar] [CrossRef]
- Rømer, J.; Lund, L.R.; Eriksen, J.; Pyke, C.; Kristensen, P.; Danø, K. The receptor for urokinase-type plasminogen activator is expressed by keratinocytes at the leading edge during re-epithelialization of mouse skin wounds. J. Invest. Dermatol. 1994, 102, 519–522. [Google Scholar] [CrossRef]
- Laerum, O.D.; Illemann, M.; Skarstein, A.; Helgeland, L.; Ovrebø, K.; Danø, K.; Nielsen, B.S. Crohn’s disease but not chronic ulcerative colitis induces the expression of PAI-1 in enteric neurons. Am. J. Gastroenterol. 2008, 103, 2350–2358. [Google Scholar] [CrossRef]
- Busso, N.; Péclat, V.; So, A.; Sappino, A.P. Plasminogen activation in synovial tissues: Differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann. Rheum. Dis. 1997, 56, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Thornton, S.; Raghu, H.; Cruz, C.; Frederick, M.D.; Palumbo, J.S.; Mullins, E.S.; Almholt, K.; Usher, P.A.; Flick, M.J. Urokinase plasminogen activator and receptor promote collagen-induced arthritis through expression in hematopoietic cells. Blood Adv. 2017, 1, 545–556. [Google Scholar] [CrossRef]
- Almholt, K.; Hebsgaard, J.B.; Nansen, A.; Andersson, C.; Pass, J.; Rønø, B.; Thygesen, P.; Pelzer, H.; Loftager, M.; Lund, I.K.; et al. Antibody-Mediated Neutralization of uPA Proteolytic Function Reduces Disease Progression in Mouse Arthritis Models. J. Immunol. 2018, 200, 957–965. [Google Scholar] [CrossRef]
- Cai, X.; Carlson, J.; Stoicov, C.; Li, H.; Wang, T.C.; Houghton, J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 2005, 128, 1937–1952. [Google Scholar] [CrossRef]
- Karam, S.M.; Leblond, C.P. Dynamics of epithelial cells in the corpus of the mouse stomach. II. Outward migration of pit cells. Anat. Rec. 1993, 236, 280–296. [Google Scholar] [CrossRef]
- Lee, A.; O’Rourke, J.; De Ungria, M.C.; Robertson, B.; Daskalopoulos, G.; Dixon, M.F. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology 1997, 112, 1386–1397. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Ferrero, R.L.; Kusters, J.G. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 2002, 7, 139–140; [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.A.; Kersulyte, D.; Mefford, M.; Danon, S.J.; Krakowka, S.; Berg, D.E. Role of Helicobacter pylori cag region genes in colonization and gastritis in two animal models. Infect. Immun. 2001, 69, 2902–2908. [Google Scholar] [CrossRef]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Angstrom, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpízar-Alpízar, W.; Skindersoe, M.E.; Rasmussen, L.; Kriegbaum, M.C.; Christensen, I.J.; Lund, I.K.; Illemann, M.; Laerum, O.D.; Krogfelt, K.A.; Andersen, L.P.; et al. Helicobacter pylori Colonization Drives Urokinase Receptor (uPAR) Expression in Murine Gastric Epithelium During Early Pathogenesis. Microorganisms 2020, 8, 1019. https://doi.org/10.3390/microorganisms8071019
Alpízar-Alpízar W, Skindersoe ME, Rasmussen L, Kriegbaum MC, Christensen IJ, Lund IK, Illemann M, Laerum OD, Krogfelt KA, Andersen LP, et al. Helicobacter pylori Colonization Drives Urokinase Receptor (uPAR) Expression in Murine Gastric Epithelium During Early Pathogenesis. Microorganisms. 2020; 8(7):1019. https://doi.org/10.3390/microorganisms8071019
Chicago/Turabian StyleAlpízar-Alpízar, Warner, Mette E. Skindersoe, Lone Rasmussen, Mette C. Kriegbaum, Ib J. Christensen, Ida K. Lund, Martin Illemann, Ole D. Laerum, Karen A. Krogfelt, Leif P. Andersen, and et al. 2020. "Helicobacter pylori Colonization Drives Urokinase Receptor (uPAR) Expression in Murine Gastric Epithelium During Early Pathogenesis" Microorganisms 8, no. 7: 1019. https://doi.org/10.3390/microorganisms8071019
APA StyleAlpízar-Alpízar, W., Skindersoe, M. E., Rasmussen, L., Kriegbaum, M. C., Christensen, I. J., Lund, I. K., Illemann, M., Laerum, O. D., Krogfelt, K. A., Andersen, L. P., & Ploug, M. (2020). Helicobacter pylori Colonization Drives Urokinase Receptor (uPAR) Expression in Murine Gastric Epithelium During Early Pathogenesis. Microorganisms, 8(7), 1019. https://doi.org/10.3390/microorganisms8071019