Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
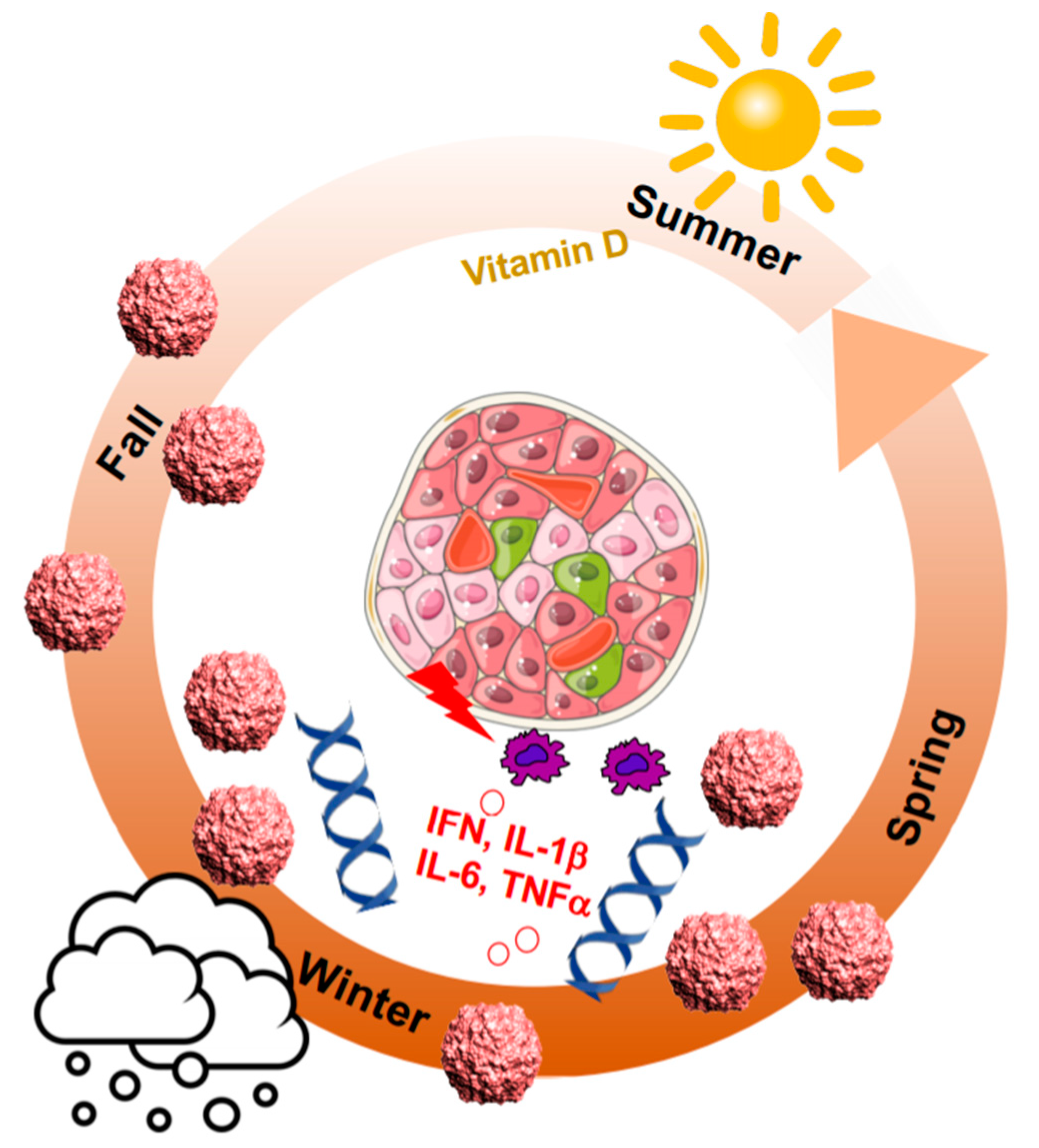
2. Seasonal Patterns of Viral and Autoimmune Diseases
3. HLA Class I and Class II Are Major Determiners for T1D
4. Direct Evidence for Enteroviral RNA in the Pancreas
5. Enteroviral Infection and T1D: Results from the TEDDY Study
6. TLR3 Signaling Leads to Enterovirus-Induced β-Cell Destruction
7. IFN-Inducible Genes Link Autoimmunity, Viral Response and β-Cell Failure in T1D
8. Why the Beta-Cell? Absence of the HIPPO Effector YAP to Balance Viral Response
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Jacobsen, L.M.; Newby, B.N.; Perry, D.J.; Posgai, A.L.; Haller, M.J.; Brusko, T.M. Immune mechanisms and pathways targeted in type 1 diabetes. Curr. Diabetes Rep. 2018, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, M.; Cooke, A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Herold, K.C.; Vignali, D.A.; Cooke, A.; Bluestone, J.A. Type 1 diabetes: Translating mechanistic observations into effective clinical outcomes. Nat. Rev. Immunol. 2013, 13, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Hyoty, H.; Taylor, K.W. The role of viruses in human diabetes. Diabetologia 2002, 45, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Yeung, W.C.; Rawlinson, W.D.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef]
- Oikarinen, M.; Tauriainen, S.; Oikarinen, S.; Honkanen, T.; Collin, P.; Rantala, I.; Maki, M.; Kaukinen, K.; Hyoty, H. Type 1 diabetes is associated with enterovirus infection in gut mucosa. Diabetes 2012, 61, 687–691. [Google Scholar] [CrossRef]
- Kyvik, K.O.; Green, A.; Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: A population based study of young Danish twins. BMJ 1995, 311, 913–917. [Google Scholar] [CrossRef]
- Adams, S.F. The seasonal variation in the onset of acute diabetes: The age and sex factor in 1,000 diabetic patients. Arch. Intern. Med. 1926, 37, 3861. [Google Scholar] [CrossRef]
- Morgan, N.G.; Richardson, S.J. Enteroviruses as causative agents in type 1 diabetes: Loose ends or lost cause? Trends Endocrinol. Metab. 2014, 25, 611–619. [Google Scholar] [CrossRef]
- Hober, D.; Alidjinou, E.K. Enteroviral pathogenesis of type 1 diabetes: Queries and answers. Curr. Opin. Infect. Dis. 2013, 26, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Hober, D.; Sane, F. Enteroviral pathogenesis of type 1 diabetes. Discov. Med. 2010, 10, 151–160. [Google Scholar] [CrossRef]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current state of the science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef] [PubMed]
- Rubino, F.; Amiel, S.A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R.H.; Mingrone, G.; Boehm, B.; Cooper, M.E.; Chai, Z.; et al. New-onset diabetes in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Chee, Y.J.; Ng, S.J.H.; Yeoh, E. Diabetic ketoacidosis precipitated by Covid-19 in a patient with newly diagnosed diabetes mellitus. Diabetes Res. Clin. Pract. 2020, 164, 108166. [Google Scholar] [CrossRef] [PubMed]
- Sin, J.; Mangale, V.; Thienphrapa, W.; Gottlieb, R.A.; Feuer, R. Recent progress in understanding coxsackievirus replication, dissemination, and pathogenesis. Virology 2015, 484, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.E. The calendar of epidemics: Seasonal cycles of infectious diseases. PLoS Pathog. 2018, 14, e1007327. [Google Scholar] [CrossRef]
- Lee, C.J.; Huang, Y.C.; Yang, S.; Tsao, K.C.; Chen, C.J.; Hsieh, Y.C.; Chiu, C.H.; Lin, T.Y. Clinical features of coxsackievirus A4, B3 and B4 infections in children. PLoS ONE 2014, 9, e87391. [Google Scholar] [CrossRef]
- Watad, A.; Azrielant, S.; Bragazzi, N.L.; Sharif, K.; David, P.; Katz, I.; Aljadeff, G.; Quaresma, M.; Tanay, G.; Adawi, M.; et al. Seasonality and autoimmune diseases: The contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun. 2017, 82, 13–30. [Google Scholar] [CrossRef]
- Moltchanova, E.V.; Schreier, N.; Lammi, N.; Karvonen, M. Seasonal variation of diagnosis of type 1 diabetes mellitus in children worldwide. Diabet. Med. 2009, 26, 673–678. [Google Scholar] [CrossRef]
- Szypowska, A.; Ramotowska, A.; Wysocka-Mincewicz, M.; Mazur, A.; Lisowicz, L.; Ben-Skowronek, I.; Sieniawska, J.; Klonowska, B.; Charemska, D.; Nawrotek, J.; et al. Seasonal variation in month of diagnosis of polish children with type 1 diabetes - A Multicenter Study. Exp. Clin. Endocrinol. Diabetes 2019, 127, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Spaans, E.A.; van Dijk, P.R.; Groenier, K.H.; Brand, P.L.; Reeser, M.H.; Bilo, H.J.; Kleefstra, N. Seasonality of diagnosis of type 1 diabetes mellitus in the Netherlands (Young Dudes-2). J. Pediatr. Endocrinol. Metab. 2016, 29, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Karuranga, S.; Salpea, P.; Saeedi, P.; Dahlquist, G.; Soltesz, G.; Ogle, G.D. Worldwide estimates of incidence, prevalence and mortality of type 1 diabetes in children and adolescents: Results from the International Diabetes Federation diabetes atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107842. [Google Scholar] [CrossRef] [PubMed]
- Kimpimaki, T.; Kupila, A.; Hamalainen, A.M.; Kukko, M.; Kulmala, P.; Savola, K.; Simell, T.; Keskinen, P.; Ilonen, J.; Simell, O.; et al. The first signs of beta-cell autoimmunity appear in infancy in genetically susceptible children from the general population: The finnish type 1 diabetes prediction and prevention study. J. Clin. Endocrinol. Metab. 2001, 86, 4782–4788. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Hansen, M.P. Type 1 diabetes associated autoimmunity. Autoimmun. Rev. 2016, 15, 644–648. [Google Scholar] [CrossRef]
- Lonnrot, M.; Korpela, K.; Knip, M.; Ilonen, J.; Simell, O.; Korhonen, S.; Savola, K.; Muona, P.; Simell, T.; Koskela, P.; et al. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: The finnish diabetes prediction and prevention study. Diabetes 2000, 49, 1314–1318. [Google Scholar] [CrossRef]
- Hiltunen, M.; Hyoty, H.; Knip, M.; Ilonen, J.; Reijonen, H.; Vahasalo, P.; Roivainen, M.; Lonnrot, M.; Leinikki, P.; Hovi, T.; et al. Islet cell antibody seroconversion in children is temporally associated with enterovirus infections. Childhood Diabetes in Finland (DiMe) Study Group. J. Infect. Dis. 1997, 175, 554–560. [Google Scholar] [CrossRef]
- Hyoty, H.; Hiltunen, M.; Knip, M.; Laakkonen, M.; Vahasalo, P.; Karjalainen, J.; Koskela, P.; Roivainen, M.; Leinikki, P.; Hovi, T.; et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 1995, 44, 652–657. [Google Scholar] [CrossRef]
- Sioofy-Khojine, A.B.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef]
- Graves, P.M.; Rotbart, H.A.; Nix, W.A.; Pallansch, M.A.; Erlich, H.A.; Norris, J.M.; Hoffman, M.; Eisenbarth, G.S.; Rewers, M. Prospective study of enteroviral infections and development of beta-cell autoimmunity. diabetes autoimmunity study in the young (DAISY). Diabetes Res. Clin. Pract. 2003, 59, 51–61. [Google Scholar] [CrossRef]
- Fuchtenbusch, M.; Irnstetter, A.; Jager, G.; Ziegler, A.G. No evidence for an association of coxsackie virus infections during pregnancy and early childhood with development of islet autoantibodies in offspring of mothers or fathers with type 1 diabetes. J. Autoimmun. 2001, 17, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C. Vitamin D and diabetes: Where do we stand? Diabetes Res. Clin. Pract. 2015, 108, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Ricordi, C.; Sanchez, J.; Clare-Salzler, M.J.; Padilla, N.; Fuenmayor, V.; Chavez, C.; Alvarez, A.; Baidal, D.; Alejandro, R.; et al. Influence of vitamin D on islet autoimmunity and beta-cell function in type 1 diabetes. Nutrients 2019, 11, 2185. [Google Scholar] [CrossRef] [PubMed]
- Dopico, X.C.; Evangelou, M.; Ferreira, R.C.; Guo, H.; Pekalski, M.L.; Smyth, D.J.; Cooper, N.; Burren, O.S.; Fulford, A.J.; Hennig, B.J.; et al. Widespread seasonal gene expression reveals annual differences in human immunity and physiology. Nat. Commun. 2015, 6, 7000. [Google Scholar] [CrossRef] [PubMed]
- Lebailly, B.; Boitard, C.; Rogner, U.C. Circadian rhythm-related genes: Implication in autoimmunity and type 1 diabetes. Diabetes Obes. Metab. 2015, 17 (Suppl. 1), 134–138. [Google Scholar] [CrossRef]
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631. [Google Scholar] [CrossRef]
- Javeed, N.; Brown, M.R.; Rakshit, K.; Her, T.; Sen, S.K.; Matveyenko, A.V. Pro-inflammatory cytokine interleukin 1beta disrupts beta cell circadian clock function and regulation of insulin secretion. Endocrinology 2020. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, D.; Liu, N.; Xiong, W.; Huang, H.; Li, Y.; Ma, Z.; Zhao, H.; Chen, P.; Qi, X.; et al. Reciprocal Regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab. 2017, 25, 73–85. [Google Scholar] [CrossRef]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc. Nat. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef]
- He, W.; Rebello, O.; Savino, R.; Terracciano, R.; Schuster-Klein, C.; Guardiola, B.; Maedler, K. TLR4 triggered complex inflammation in human pancreatic islets. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 86–97. [Google Scholar] [CrossRef]
- He, W.; Yuan, T.; Maedler, K. Macrophage-associated pro-inflammatory state in human islets from obese individuals. Nutr. Diabetes 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Peek, C.B.; Levine, D.C.; Cedernaes, J.; Taguchi, A.; Kobayashi, Y.; Tsai, S.J.; Bonar, N.A.; McNulty, M.R.; Ramsey, K.M.; Bass, J. Circadian clock interaction with HIF1alpha mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 2017, 25, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Schurman, S.H.; O’Hanlon, T.P.; McGrath, J.A.; Gruzdev, A.; Bektas, A.; Xu, H.; Garantziotis, S.; Zeldin, D.C.; Miller, F.W. Transethnic associations among immune-mediated diseases and single-nucleotide polymorphisms of the aryl hydrocarbon response gene ARNT and the PTPN22 immune regulatory gene. J. Autoimmun. 2020, 107, 102363. [Google Scholar] [CrossRef] [PubMed]
- Paroni, F.; Domsgen, E.; Maedler, K. CXCL10- a path to beta-cell death. Islets 2009, 1, 256–259. [Google Scholar] [CrossRef]
- Al Badri, A.M.; Foulis, A.K.; Todd, P.M.; Gariouch, J.J.; Gudgeon, J.E.; Stewart, D.G.; Gracie, J.A.; Goudie, R.B. Abnormal expression of MHC class II and ICAM-1 by melanocytes in vitiligo. J. Pathol. 1993, 169, 203–206. [Google Scholar] [CrossRef]
- Noble, J.A. Immunogenetics of type 1 diabetes: A comprehensive review. J. Autoimmun. 2015, 64, 101–112. [Google Scholar] [CrossRef]
- Valdes, A.M.; Erlich, H.A.; Noble, J.A. Human leukocyte antigen class I B and C loci contribute to type 1 diabetes (T1D) susceptibility and age at T1D onset. Hum. Immunol. 2005, 66, 301–313. [Google Scholar] [CrossRef]
- Nejentsev, S.; Howson, J.M.; Walker, N.M.; Szeszko, J.; Field, S.F.; Stevens, H.E.; Reynolds, P.; Hardy, M.; King, E.; Masters, J.; et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007, 450, 887–892. [Google Scholar] [CrossRef]
- Valdes, A.M.; Erlich, H.A.; Carlson, J.; Varney, M.; Moonsamy, P.V.; Noble, J.A. Use of class I and class II HLA loci for predicting age at onset of type 1 diabetes in multiple populations. Diabetologia 2012, 55, 2394–2401. [Google Scholar] [CrossRef]
- Hietala, K.; Harjutsalo, V.; Forsblom, C.; Summanen, P.; Groop, P.H.; FinnDiane Study, G. Age at onset and the risk of proliferative retinopathy in type 1 diabetes. Diabetes Care 2010, 33, 1315–1319. [Google Scholar] [CrossRef]
- Hoffmann, V.S.; Weiss, A.; Winkler, C.; Knopff, A.; Jolink, M.; Bonifacio, E.; Ziegler, A.G. Landmark models to define the age-adjusted risk of developing stage 1 type 1 diabetes across childhood and adolescence. BMC Med. 2019, 17, 125. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Calvo, T.; Richardson, S.J.; Pugliese, A. pancreas pathology during the natural history of type 1 diabetes. Curr. Diabetes Rep. 2018, 18, 124. [Google Scholar] [CrossRef]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jorgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Chehadeh, W.; Kerr-Conte, J.; Pattou, F.; Alm, G.; Lefebvre, J.; Wattre, P.; Hober, D. Persistent infection of human pancreatic islets by coxsackievirus B is associated with alpha interferon synthesis in beta cells. J. Virol. 2000, 74, 10153–10164. [Google Scholar] [CrossRef]
- Vives-Pi, M.; Armengol, M.P.; Alcalde, L.; Costa, M.; Somoza, N.; Vargas, F.; Jaraquemada, D.; Pujol-Borrell, R. Expression of transporter associated with antigen processing-1 in the endocrine cells of human pancreatic islets: Effect of cytokines and evidence of hyperexpression in IDDM. Diabetes 1996, 45, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op de Beeck, A.; Coomans de Brachene, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Kahrs, C.R.; Chuda, K.; Tapia, G.; Stene, L.C.; Marild, K.; Rasmussen, T.; Ronningen, K.S.; Lundin, K.E.A.; Kramna, L.; Cinek, O.; et al. Enterovirus as trigger of coeliac disease: Nested case-control study within prospective birth cohort. BMJ 2019, 364, l231. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, A.; Donnachie, E.; Jergens, S.; Ziegler, A.G. Infections in early life and development of type 1 diabetes. Jama 2016, 315, 1899–1901. [Google Scholar] [CrossRef]
- Gamble, D.R.; Kinsley, M.L.; FitzGerald, M.G.; Bolton, R.; Taylor, K.W. Viral antibodies in diabetes mellitus. Br. Med. J. 1969, 3, 627–630. [Google Scholar] [CrossRef]
- Domsgen, E.; Paroni, F.; Kerr-Conte, J.; Dotzauer, A.; Maedler, K. Virus-induced beta cell death depends on CXCL10 and the AKT-JNK-PKR crosstalk. Diabetologia 2010, 53, S186. [Google Scholar]
- Christen, U.; von Herrath, M.G. Do viral infections protect from or enhance type 1 diabetes and how can we tell the difference? Cell Mol. Immunol. 2011, 8, 193–198. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T.; von Herrath, M.G. Enterovirus infection and type 1 diabetes: Closing in on a link? Diabetes 2015, 64, 1503–1505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoon, J.W.; Austin, M.; Onodera, T.; Notkins, A.L. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 1979, 300, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Skog, O.; Ingvast, S.; Korsgren, O. Evaluation of RT-PCR and immunohistochemistry as tools for detection of enterovirus in the human pancreas and islets of Langerhans. J. Clin. Virol. 2014, 61, 242–247. [Google Scholar] [CrossRef]
- Richardson, S.J.; Leete, P.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia 2013, 56, 185–193. [Google Scholar] [CrossRef]
- Oikarinen, S.; Tauriainen, S.; Hober, D.; Lucas, B.; Vazeou, A.; Sioofy-Khojine, A.; Bozas, E.; Muir, P.; Honkanen, H.; Ilonen, J.; et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 2014, 63, 655–662. [Google Scholar] [CrossRef]
- Dezayee, Z.M. The status of serum gamma-interferonand antiviral antibodies in patients with type I and type 2 diabetes: A comparative study. J. Res. Med. Sci. 2012, 17, 855–858. [Google Scholar]
- Alberti, A.M.; Amato, C.; Candela, A.; Costantino, F.; Grandolfo, M.E.; Lombardi, F.; Novello, F.; Orsini, M.; Santoro, R. Serum antibodies against Coxsackie B1-6 viruses in type 1 diabetics. Acta Diabetol. Lat. 1985, 22, 33–38. [Google Scholar] [CrossRef]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyoty, H. Enterovirus RNA in blood is linked to the development of type 1 diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef]
- Genoni, A.; Canducci, F.; Rossi, A.; Broccolo, F.; Chumakov, K.; Bono, G.; Salerno-Uriarte, J.; Salvatoni, A.; Pugliese, A.; Toniolo, A. Revealing enterovirus infection in chronic human disorders: An integrated diagnostic approach. Sci. Rep. 2017, 7, 5013. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Horton, J.L.; Pang, C.N.I.; Jain, K.; Leung, P.; Isaacs, S.R.; Bull, R.A.; Luciani, F.; Wilkins, M.R.; Catteau, J.; et al. Higher abundance of enterovirus A species in the gut of children with islet autoimmunity. Sci. Rep. 2019, 9, 1749. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Jun, H.S. Viruses cause type 1 diabetes in animals. Ann. N. Y. Acad. Sci. 2006, 1079, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hober, D.; Sane, F. Enteroviruses and type 1 diabetes. BMJ 2011, 342, c7072. [Google Scholar] [CrossRef]
- Sane, F.; Caloone, D.; Gmyr, V.; Engelmann, I.; Belaich, S.; Kerr-Conte, J.; Pattou, F.; Desailloud, R.; Hober, D. Coxsackievirus B4 can infect human pancreas ductal cells and persist in ductal-like cell cultures which results in inhibition of Pdx1 expression and disturbed formation of islet-like cell aggregates. Cell. Mol. Life Sci. 2013, 70, 4169–4180. [Google Scholar] [CrossRef]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef]
- Busse, N.; Paroni, F.; Richardson, S.J.; Laiho, J.E.; Oikarinen, M.; Frisk, G.; Hyoty, H.; de Koning, E.; Morgan, N.G.; Maedler, K. Detection and localization of viral infection in the pancreas of patients with type 1 diabetes using short fluorescently-labelled oligonucleotide probes. Oncotarget 2017, 8, 12620–12636. [Google Scholar] [CrossRef]
- Laiho, J.E.; Oikarinen, S.; Oikarinen, M.; Larsson, P.G.; Stone, V.M.; Hober, D.; Oberste, S.; Flodstrom-Tullberg, M.; Isola, J.; Hyoty, H. Application of bioinformatics in probe design enables detection of enteroviruses on different taxonomic levels by advanced in situ hybridization technology. J. Clin. Virol. 2015, 69, 165–171. [Google Scholar] [CrossRef]
- Laiho, J.E.; Oikarinen, M.; Richardson, S.J.; Frisk, G.; Nyalwidhe, J.; Burch, T.C.; Morris, M.A.; Oikarinen, S.; Pugliese, A.; Dotta, F.; et al. Relative sensitivity of immunohistochemistry, multiple reaction monitoring mass spectrometry, in situ hybridization and PCR to detect Coxsackievirus B1 in A549 cells. J. Clin. Virol. 2016, 77, 21–28. [Google Scholar] [CrossRef]
- Geravandi, S.; Maedler, K. Enteroviral mRNA detection in the pancreas of patients with type 1 diabetes. Diabetol. 2019, 62, S7. [Google Scholar]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef]
- Battaglia, M.; Atkinson, M.A. The streetlight effect in type 1 diabetes. Diabetes 2015, 64, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Leete, P.; Dhayal, S.; Russell, M.A.; Oikarinen, M.; Laiho, J.E.; Svedin, E.; Lind, K.; Rosenling, T.; Chapman, N.; et al. Evaluation of the fidelity of immunolabelling obtained with clone 5D8/1, a monoclonal antibody directed against the enteroviral capsid protein, VP1, in human pancreas. Diabetologia 2014, 57, 392–401. [Google Scholar] [CrossRef]
- Hansson, S.F.; Korsgren, S.; Ponten, F.; Korsgren, O. Enteroviruses and the pathogenesis of type 1 diabetes revisited: Cross-reactivity of enterovirus capsid protein (VP1) antibodies with human mitochondrial proteins. J. Pathol. 2013, 229, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Mena, I.; Fischer, C.; Gebhard, J.R.; Perry, C.M.; Harkins, S.; Whitton, J.L. Coxsackievirus infection of the pancreas: Evaluation of receptor expression, pathogenesis, and immunopathology. Virology 2000, 271, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.J.; Saunders, D.C.; Dai, C.; Poffenberger, G.; Cairns, B.; Serreze, D.V.; Harlan, D.M.; Bottino, R.; Brissova, M.; Powers, A.C. Decreased pancreatic acinar cell number in type 1 diabetes. Diabetologia 2020. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.L.; Filipp, S.L.; Grajo, J.R.; Nambam, B.; Beegle, R.; Middlebrooks, E.H.; Gurka, M.J.; Atkinson, M.A.; Schatz, D.A.; Haller, M.J. Relative pancreas volume is reduced in first-degree relatives of patients with type 1 diabetes. Diabetes Care 2019, 42, 281–287. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.L.; Kaddis, J.S.; Wasserfall, C.; Haller, M.J.; Pugliese, A.; Schatz, D.A.; Shuster, J.J.; Atkinson, M.A. The influence of type 1 diabetes on pancreatic weight. Diabetologia 2016, 59, 217–221. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvetnick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef]
- Sane, F.; Moumna, I.; Hober, D. Group B coxsackieviruses and autoimmunity: Focus on type 1 diabetes. Expert Rev. Clin. Immunol. 2011, 7, 357–366. [Google Scholar] [CrossRef]
- Zarozinski, C.C.; Welsh, R.M. Minimal bystander activation of CD8 T cells during the virus-induced polyclonal T cell response. J. Exp. Med. 1997, 185, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Seewaldt, S.; Thomas, H.E.; Ejrnaes, M.; Christen, U.; Wolfe, T.; Rodrigo, E.; Coon, B.; Michelsen, B.; Kay, T.W.; von Herrath, M.G. Virus-induced autoimmune diabetes: Most beta-cells die through inflammatory cytokines and not perforin from autoreactive (anti-viral) cytotoxic T-lymphocytes. Diabetes 2000, 49, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Bogdani, M.; Korpos, E.; Simeonovic, C.J.; Parish, C.R.; Sorokin, L.; Wight, T.N. Extracellular matrix components in the pathogenesis of type 1 diabetes. Curr. Diabetes Rep. 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef]
- Domsgen, E.; Paroni, F.; Kerr-Conte, J.; Dotzauer, A.; Maedler, K. Coxsackievirus initiates strong immune response and death of beta cells. Diabetologia 2012, 55, S196–S197. [Google Scholar]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Guo, H.; Coulson, R.M.; Smyth, D.J.; Pekalski, M.L.; Burren, O.S.; Cutler, A.J.; Doecke, J.D.; Flint, S.; McKinney, E.F.; et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 2014, 63, 2538–2550. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. Beta-cell autophagy in diabetes pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef]
- Kim, K.S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.K.; Kim, K.; Barry, W.H.; Chapman, N.M. 5′-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef]
- Lindfors, K.; Lin, J.; Lee, H.S.; Hyoty, H.; Nykter, M.; Kurppa, K.; Liu, E.; Koletzko, S.; Rewers, M.; Hagopian, W.; et al. Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: The TEDDY study. Gut 2019. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Aida, K.; Nishida, Y.; Tanaka, S.; Maruyama, T.; Shimada, A.; Awata, T.; Suzuki, M.; Shimura, H.; Takizawa, S.; Ichijo, M.; et al. RIG-I- and MDA5-initiated innate immunity linked with adaptive immunity accelerates beta-cell death in fulminant type 1 diabetes. Diabetes 2011, 60, 884–889. [Google Scholar] [CrossRef]
- Lee, A.S.; Ghoreishi, M.; Cheng, W.K.; Chang, T.Y.; Zhang, Y.Q.; Dutz, J.P. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia 2011, 54, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef]
- Smyth, D.J.; Cooper, J.D.; Bailey, R.; Field, S.; Burren, O.; Smink, L.J.; Guja, C.; Ionescu-Tirgoviste, C.; Widmer, B.; Dunger, D.B.; et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006, 38, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Brondani Lde, A.; Bauer, A.C.; Canani, L.H.; Crispim, D. Polymorphisms in the TLR3 gene are associated with risk for type 1 diabetes mellitus. Eur. J. Endocrinol. 2014, 170, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Morse, Z.J.; Horwitz, M.S. Innate viral receptor signaling determines type 1 diabetes Onset. Front. Endocrinol. 2017, 8, 249. [Google Scholar] [CrossRef]
- Tai, N.; Wong, F.S.; Wen, L. The role of the innate immune system in destruction of pancreatic beta cells in NOD mice and humans with type I diabetes. J. Autoimmun. 2016, 71, 26–34. [Google Scholar] [CrossRef]
- Pirie, F.J.; Pegoraro, R.; Motala, A.A.; Rauff, S.; Rom, L.; Govender, T.; Esterhuizen, T.M. Toll-like receptor 3 gene polymorphisms in South African Blacks with type 1 diabetes. Tissue Antigens 2005, 66, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Fichna, M.; Zurawek, M.; Fichna, P.; Januszkiewicz-Lewandowska, D.; Ruchala, M.; Nowak, J. Polymorphisms of the toll-like receptor-3 gene in autoimmune adrenal failure and type 1 diabetes in Polish patients. Arch. Immunol. Ther. Exp. 2016, 64, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, S.; Imagawa, A.; Tauriainen, S.; Iino, M.; Oikarinen, M.; Abiru, H.; Tamaki, K.; Seino, H.; Nishi, K.; Takase, I.; et al. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr. J. 2010, 57, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Huhn, M.H.; Wolf, M.; Olsson, A.; Jacobson, S.; Williams, B.R.; Korsgren, O.; Flodstrom-Tullberg, M. Interferons induce an antiviral state in human pancreatic islet cells. Virology 2007, 367, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Domsgen, E.; Paroni, F.; Kerr-Conte, J.; Dotzauer, A.; Maedler, K. Endosomal TLR3 activation by Coxsackievirus initiates strong immune response and death of beta cells. Diabetologia 2011, 54, S184. [Google Scholar]
- Berg, A.K.; Korsgren, O.; Frisk, G. Induction of the chemokine interferon-gamma-inducible protein-10 in human pancreatic islets during enterovirus infection. Diabetologia 2006, 49, 2697–2703. [Google Scholar] [CrossRef]
- Christen, U.; McGavern, D.B.; Luster, A.D.; von Herrath, M.G.; Oldstone, M.B. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol. 2003, 171, 6838–6845. [Google Scholar] [CrossRef]
- Richer, M.J.; Lavallee, D.J.; Shanina, I.; Horwitz, M.S. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS ONE 2009, 4, e4127. [Google Scholar] [CrossRef]
- McCartney, S.A.; Vermi, W.; Lonardi, S.; Rossini, C.; Otero, K.; Calderon, B.; Gilfillan, S.; Diamond, M.S.; Unanue, E.R.; Colonna, M. RNA sensor-induced type I IFN prevents diabetes caused by a beta cell-tropic virus in mice. J. Clin. Investig. 2011, 121, 1497–1507. [Google Scholar] [CrossRef]
- Wong, F.S.; Hu, C.; Zhang, L.; Du, W.; Alexopoulou, L.; Flavell, R.A.; Wen, L. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann. N. Y. Acad. Sci. 2008, 1150, 146–148. [Google Scholar] [CrossRef]
- McCall, K.D.; Thuma, J.R.; Courreges, M.C.; Benencia, F.; James, C.B.; Malgor, R.; Kantake, N.; Mudd, W.; Denlinger, N.; Nolan, B.; et al. Toll-like receptor 3 is critical for coxsackievirus B4-induced type 1 diabetes in female NOD mice. Endocrinology 2015, 156, 453–461. [Google Scholar] [CrossRef]
- Gulden, E.; Chao, C.; Tai, N.; Pearson, J.A.; Peng, J.; Majewska-Szczepanik, M.; Zhou, Z.; Wong, F.S.; Wen, L. TRIF deficiency protects non-obese diabetic mice from type 1 diabetes by modulating the gut microbiota and dendritic cells. J. Autoimmun. 2018, 93, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Spagnuolo, I.; Patti, A.; Grieco, F.A.; Cataldo, D.; Ferretti, E.; Tiberti, C.; Dotta, F. MicroRNA expression fingerprint in serum of type 1 diabetic patients. Diabetologia 2012, 55, S48. [Google Scholar]
- O’Neill, L.A.; Sheedy, F.J.; McCoy, C.E. MicroRNAs: The fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 2011, 11, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Storling, J.; Pociot, F. Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes 2017, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Nyaga, D.M.; Vickers, M.H.; Jefferies, C.; Perry, J.K.; O’Sullivan, J.M. Type 1 diabetes mellitus-associated genetic variants contribute to overlapping immune regulatory networks. Front. Genet. 2018, 9, 535. [Google Scholar] [CrossRef]
- Looney, B.M.; Xia, C.Q.; Concannon, P.; Ostrov, D.A.; Clare-Salzler, M.J. Effects of type 1 diabetes-associated IFIH1 polymorphisms on MDA5 function and expression. Curr. Diabetes Rep. 2015, 15, 96. [Google Scholar] [CrossRef]
- Kallionpaa, H.; Elo, L.L.; Laajala, E.; Mykkanen, J.; Ricano-Ponce, I.; Vaarma, M.; Laajala, T.D.; Hyoty, H.; Ilonen, J.; Veijola, R.; et al. Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 2014, 63, 2402–2414. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Dotta, F.; Amirian, N.; Campbell, P.D.; Kay, T.W.; Atkinson, M.A.; Roep, B.O.; von Herrath, M.G. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med. 2012, 209, 51–60. [Google Scholar] [CrossRef]
- Colli, M.L.; Moore, F.; Gurzov, E.N.; Ortis, F.; Eizirik, D.L. MDA5 and PTPN2, two candidate genes for type 1 diabetes, modify pancreatic beta-cell responses to the viral by-product double-stranded RNA. Hum. Mol. Genet. 2010, 19, 135–146. [Google Scholar] [CrossRef]
- Domsgen, E.; Lind, K.; Kong, L.; Huhn, M.H.; Rasool, O.; van Kuppeveld, F.; Korsgren, O.; Lahesmaa, R.; Flodstrom-Tullberg, M. An IFIH1 gene polymorphism associated with risk for autoimmunity regulates canonical antiviral defence pathways in Coxsackievirus infected human pancreatic islets. Sci. Rep. 2016, 6, 39378. [Google Scholar] [CrossRef]
- Duan, S.; Paulson, J.C. Siglecs as immune cell checkpoints in disease. Annu. Rev. Immunol. 2020, 38, 365–395. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, G.; Stolz, K.; Hauke, M.; Morgan, N.G.; Varki, A.; de Koning, E.; Kelm, S.; Maedler, K. Siglec-7 restores beta-cell function and survival and reduces inflammation in pancreatic islets from patients with diabetes. Sci. Rep. 2017, 7, 45319. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. TANK-binding kinase 1 as a novel therapeutic target for viral diseases. Expert Opin. Ther. Targets 2019, 23, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, F.; Chen, S.; Plouffe, S.W.; Wu, S.; Liu, S.; Li, X.; Zhou, R.; Wang, J.; Zhao, B.; et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 2017, 19, 362–374. [Google Scholar] [CrossRef]
- Munoz-Wolf, N.; Lavelle, E.C. Hippo interferes with antiviral defences. Nat. Cell Biol. 2017, 19, 267–269. [Google Scholar] [CrossRef]
- Meng, F.; Zhou, R.; Wu, S.; Zhang, Q.; Jin, Q.; Zhou, Y.; Plouffe, S.W.; Liu, S.; Song, H.; Xia, Z.; et al. Mst1 shuts off cytosolic antiviral defense through IRF3 phosphorylation. Genes Dev. 2016, 30, 1086–1100. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef]
- Gao, T.; Zhou, D.; Yang, C.; Singh, T.; Penzo-Mendez, A.; Maddipati, R.; Tzatsos, A.; Bardeesy, N.; Avruch, J.; Stanger, B.Z. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology 2013, 144, 1543–1553, 1553.e1. [Google Scholar] [CrossRef]
- George, N.M.; Boerner, B.P.; Mir, S.U.; Guinn, Z.; Sarvetnick, N.E. Exploiting Expression of Hippo effector, yap, for expansion of functional islet mass. Mol. Endocrinol. 2015, 29, 1594–1607. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Mizrachi, E.B.; Ocana, A.G.; Stewart, A.F. Human beta-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Rafizadeh, S.; Azizi, A.; Lupse, B.; Gorrepati, K.; Awal, S.; Oberholzer, J.; Maedler, K.; Ardestani, A. Proproliferative and antiapoptotic action of exogenously introduced YAP in pancreatic β cells. JCI Insight 2016, 1, e86326. [Google Scholar] [CrossRef] [PubMed]
- Blodgett, D.M.; Nowosielska, A.; Afik, S.; Pechhold, S.; Cura, A.J.; Kennedy, N.J.; Kim, S.; Kucukural, A.; Davis, R.J.; Kent, S.C.; et al. Novel observations from Next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 2015, 64, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Benner, C.; van der Meulen, T.; Caceres, E.; Tigyi, K.; Donaldson, C.J.; Huising, M.O. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC Genom. 2014, 15, 620. [Google Scholar] [CrossRef]
- Pullen, T.J.; Khan, A.M.; Barton, G.; Butcher, S.A.; Sun, G.; Rutter, G.A. Identification of genes selectively disallowed in the pancreatic islet. Islets 2010, 2, 89–95. [Google Scholar] [CrossRef]
- Pullen, T.J.; Huising, M.O.; Rutter, G.A. Analysis of purified pancreatic islet beta and alpha cell transcriptomes reveals 11beta-hydroxysteroid dehydrogenase (Hsd11b1) as a novel disallowed gene. Front. Genet. 2017, 8, 41. [Google Scholar] [CrossRef]
- Ardestani, A.; Paroni, F.; Azizi, Z.; Kaur, S.; Khobragade, V.; Yuan, T.; Frogne, T.; Tao, W.; Oberholzer, J.; Pattou, F.; et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 2014, 20, 385–397. [Google Scholar] [CrossRef]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jorgensen, K.; Flodstrom-Tullberg, M.; Hyoty, H.; Insel, R.A.; Lernmark, A.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.M.; Wang, X.; Chen, Y.G.; Jia, S.; Kaldunski, M.L.; Greenbaum, C.J.; Type 1 Diabetes Trialnet Canakinumab Study Group; Mandrup-Poulsen, T.; AIDA Study Group; Hessner, M.J. Interleukin-1 antagonism moderates the inflammatory state associated with Type 1 diabetes during clinical trials conducted at disease onset. Eur. J. Immunol. 2016, 46, 1030–1046. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, A.; Li, S.; Annamalai, K.; Lupse, B.; Geravandi, S.; Dobrowolski, A.; Yu, S.; Zhu, S.; Baguley, T.D.; Surakattula, M.; et al. Neratinib protects pancreatic beta cells in diabetes. Nat. Commun. 2019, 10, 5015. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms 2020, 8, 1017. https://doi.org/10.3390/microorganisms8071017
Geravandi S, Liu H, Maedler K. Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms. 2020; 8(7):1017. https://doi.org/10.3390/microorganisms8071017
Chicago/Turabian StyleGeravandi, Shirin, Huan Liu, and Kathrin Maedler. 2020. "Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D" Microorganisms 8, no. 7: 1017. https://doi.org/10.3390/microorganisms8071017
APA StyleGeravandi, S., Liu, H., & Maedler, K. (2020). Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms, 8(7), 1017. https://doi.org/10.3390/microorganisms8071017