Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Cells and Virus Strains
2.3. Expression of EV Receptors in the AV3 Cell Line
2.4. Detection of Enteroviruses in nPOD Spleen Samples
2.5. Immune Transcriptome of Control Uninfected and Infected AV3 Cells
2.6. Release of Cytokines by Uninfected and Infected AV3 Cell Cultures
2.7. Statistical Analysis
3. Results
3.1. Network of Pancreas Organ Donors with Diabetes (nPOD): Isolation of Defective Enterovirus Strains from Spleen Samples
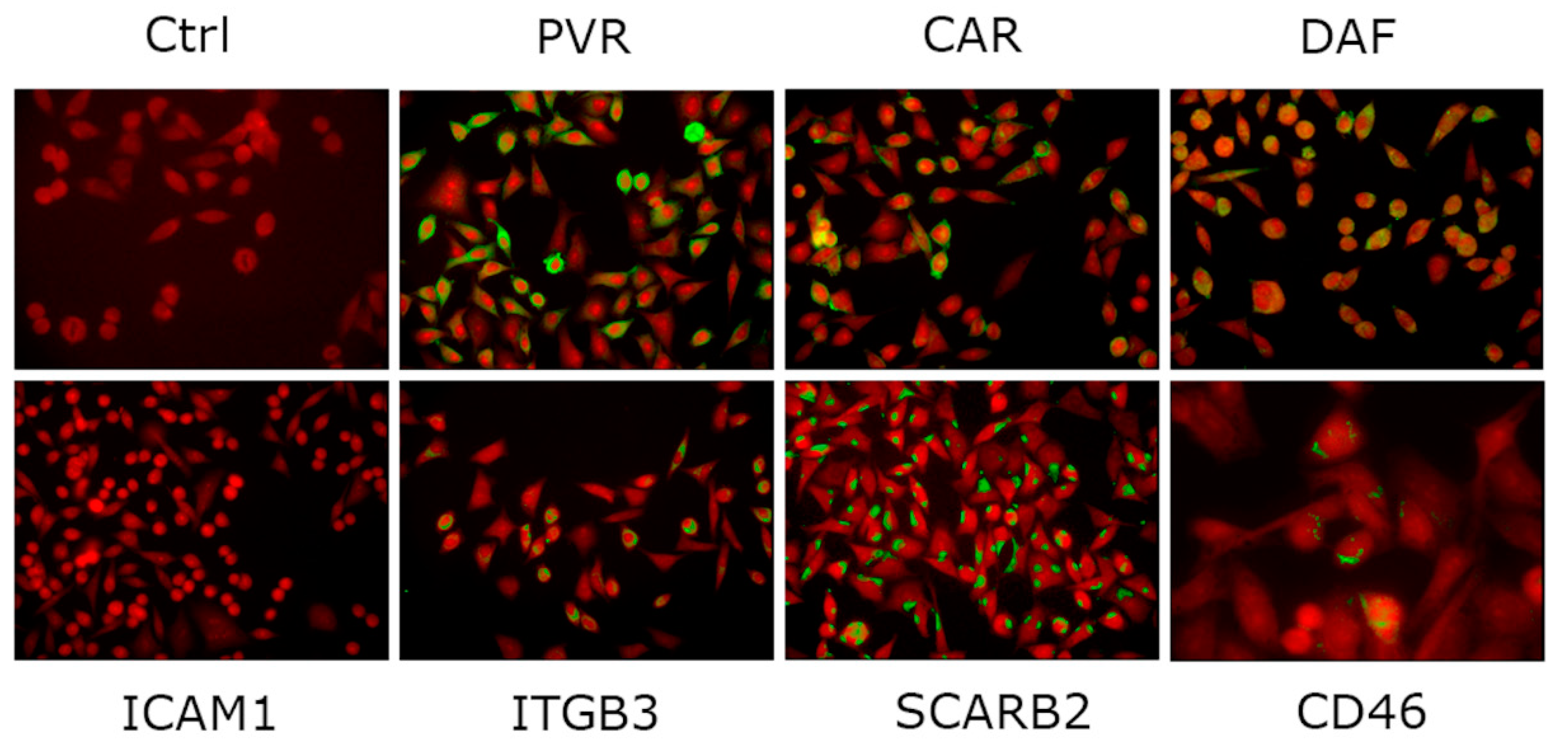
3.2. Enterovirus Receptors in the AV3 Cell Line
3.3. Infection of AV3 Cells with Replication-Competent CBV3 and with Defective EV Isolates from T1D Cases
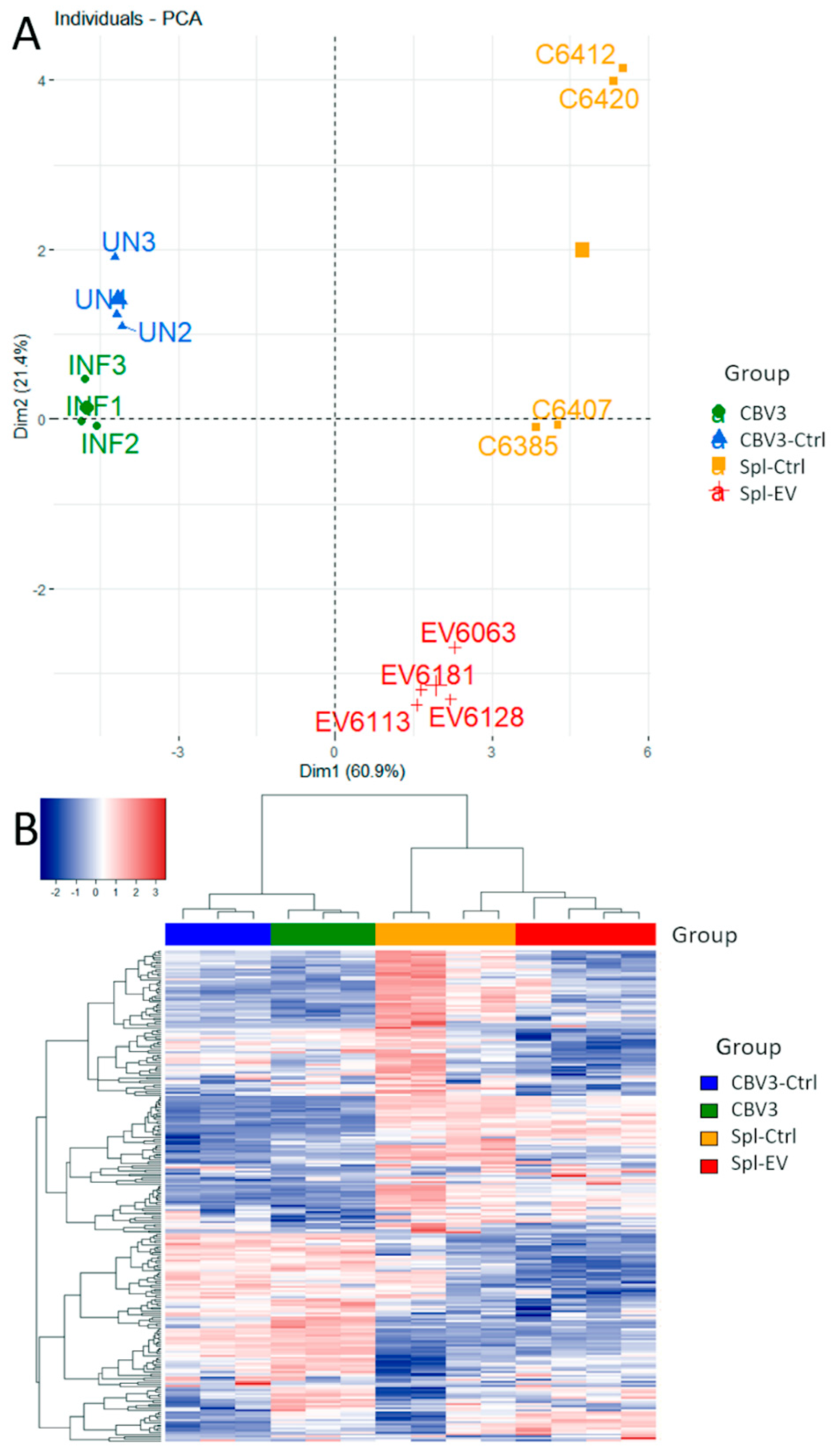
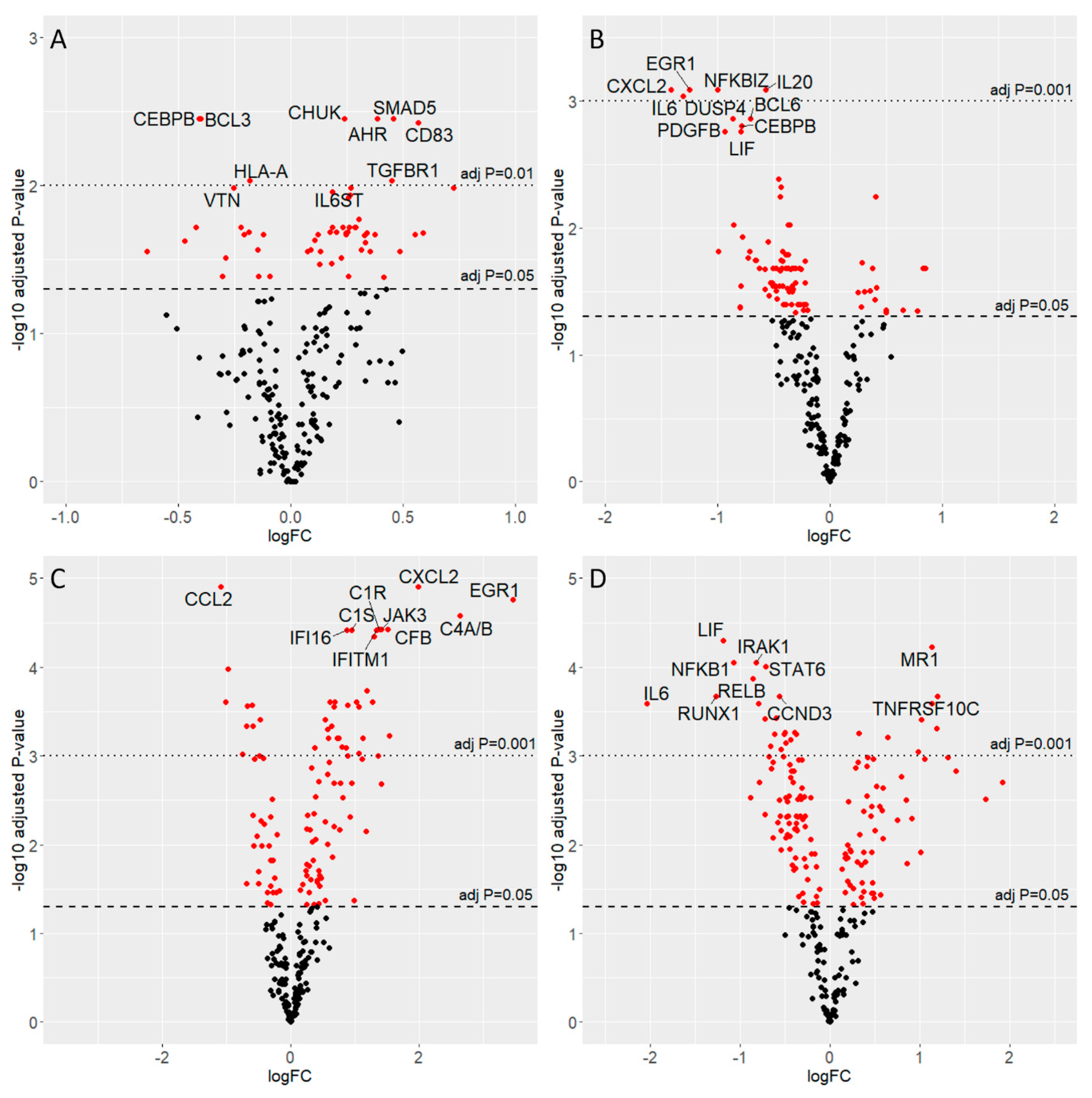
3.4. Gene Expression Analysis
3.5. Interferon Pathways: Gene Transcription Changes in the Acute and the Slow Enterovirus Infection Models
3.6. Pathways Activated in the Acute and Slow Models of Infection
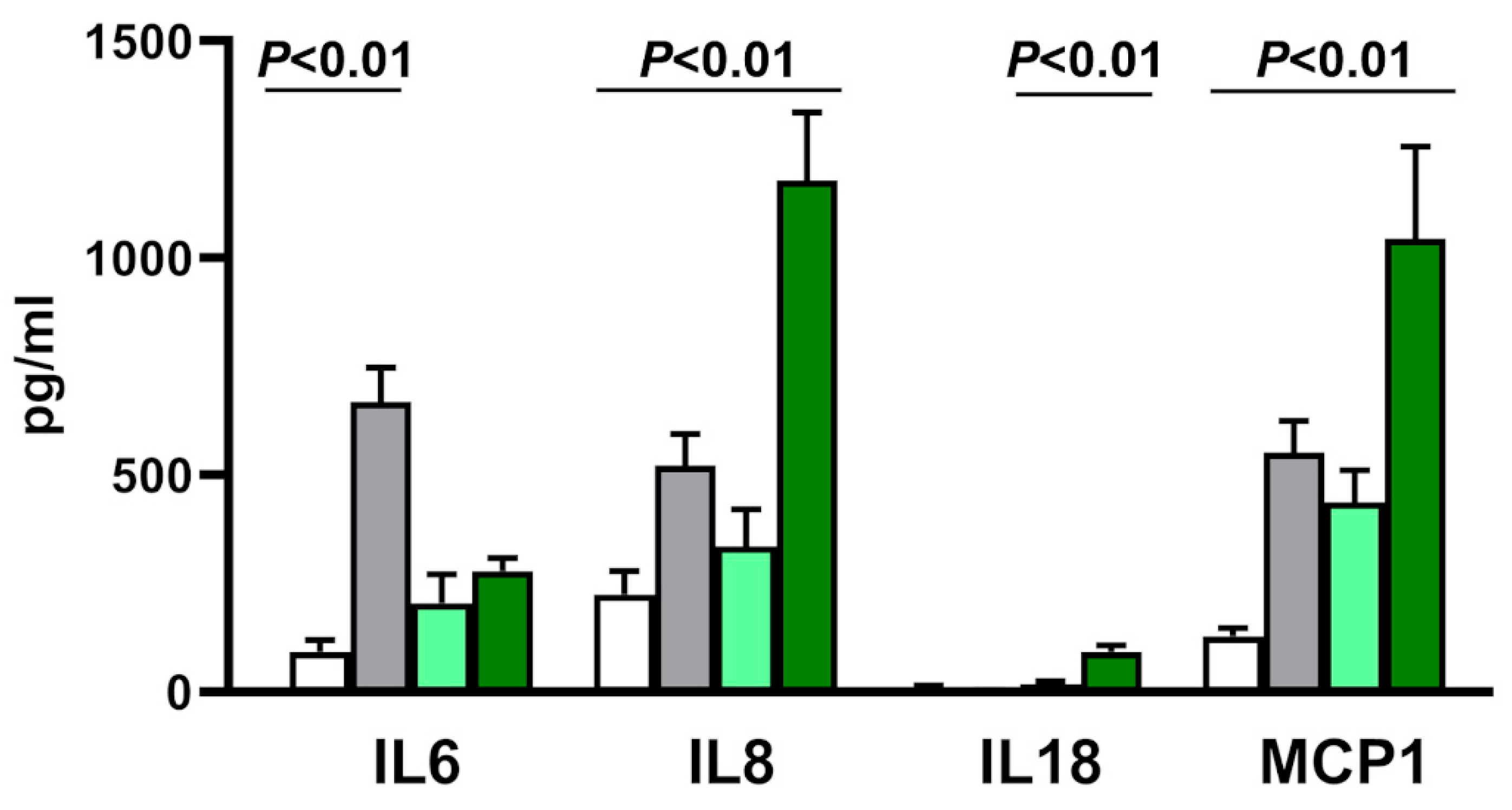
3.7. Cytokine Transcription and Secretion in Acute and Slow Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richardson, S.J.; Morgan, N.G. Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharmacol. 2018, 43, 11–19. [Google Scholar] [CrossRef]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a low-grade enteroviral infection in the islets of langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef]
- Sioofy-Khojine, A.-B.; Lehtonen, J.; Nurminen, N.; Laitinen, O.H.; Oikarinen, S.; Huhtala, H.; Pakkanen, O.; Ruokoranta, T.; Hankaniemi, M.M.; Toppari, J.; et al. Coxsackievirus B1 infections are associated with the initiation of insulin-driven autoimmunity that progresses to type 1 diabetes. Diabetologia 2018, 61, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, H.; Oikarinen, S.; Nurminen, N.; Laitinen, O.H.; Huhtala, H.; Lehtonen, J.; Ruokoranta, T.; Hankaniemi, M.M.; Lecouturier, V.; Almond, J.W.; et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: Possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia 2017, 60, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Hyöty, H. Viruses in type 1 diabetes. Pediatr. Diabetes 2016, 17 (Suppl. 22), 56–64. [Google Scholar] [CrossRef]
- Hyöty, H.; Leon, F.; Knip, M. Developing a vaccine for type 1 diabetes by targeting coxsackievirus B. Expert Rev. Vaccines 2018, 17, 1071–1083. [Google Scholar] [CrossRef]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jørgensen, K.; Flodström-Tullberg, M.; Hyöty, H.; Insel, R.A.; Lernmark, Å.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Oikarinen, M.; Laiho, J.E.; Oikarinen, S.; Richardson, S.J.; Kusmartseva, I.; Campbell-Thompson, M.; Morgan, N.G.; Pugliese, A.; Tauriainen, S.; Toniolo, A.; et al. Detection of enterovirus protein and RNA in multiple tissues from nPOD organ donors with type 1 diabetes. bioRxiv 2018. [Google Scholar] [CrossRef]
- Massilamany, C.; Koenig, A.; Reddy, J.; Huber, S.; Buskiewicz, I. Autoimmunity in picornavirus infections. Curr. Opin. Virol. 2016, 16, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells from Donors with Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeus, E.; De Neuter, N.; Lemay, A.; Pattyn, L.; Tuerlinckx, D.; Weynants, D.; Van Lede, K.; van Berlaer, G.; Bulckaert, D.; Boiy, T.; et al. Diagnosing enterovirus meningitis via blood transcriptomics: An alternative for lumbar puncture? J. Transl. Med. 2019, 17, 282. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Holmes, C.W. Acute and chronic disease caused by enteroviruses. Virulence 2017, 8, 1062–1065. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Genoni, A.; Canducci, F.; Rossi, A.; Broccolo, F.; Chumakov, K.; Bono, G.; Salerno-Uriarte, J.; Salvatoni, A.; Pugliese, A.; Toniolo, A. Revealing enterovirus infection in chronic human disorders: An integrated diagnostic approach. Sci. Rep. 2017, 7, 5013. [Google Scholar] [CrossRef]
- Holmes, A.C.; Semler, B.L. Picornaviruses and RNA Metabolism: Local and Global Effects of Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Wan, X.; Vomund, A.N.; Peterson, O.J.; Chervonsky, A.V.; Lichti, C.F.; Unanue, E.R. The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat. Immunol. 2020, 21, 455–463. [Google Scholar] [CrossRef]
- Dasoveanu, D.C.; Park, H.J.; Ly, C.L.; Shipman, W.D.; Chyou, S.; Kumar, V.; Tarlinton, D.; Ludewig, B.; Mehrara, B.J.; Lu, T.T. Lymph node stromal CCL2 limits antibody responses. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Kaddis, J.S.; Pugliese, A.; Atkinson, M.A. A run on the biobank: What have we learned about type 1 diabetes from the nPOD tissue repository? Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 290–295. [Google Scholar] [CrossRef]
- Toniolo, A.; Onodera, T.; Jordan, G.; Yoon, J.W.; Notkins, A.L. Virus-induced diabetes mellitus. Glucose abnormalities produced in mice by the six members of the Coxsackie B virus group. Diabetes 1982, 31, 496–499. [Google Scholar] [CrossRef]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Burg, A.R.; Das, S.; Padgett, L.E.; Koenig, Z.E.; Tse, H.M. Superoxide Production by NADPH Oxidase Intensifies Macrophage Antiviral Responses during Diabetogenic Coxsackievirus Infection. J. Immunol. 2018, 200, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Oikarinen, S.; Tauriainen, S.; Hober, D.; Lucas, B.; Vazeou, A.; Sioofy-Khojine, A.; Bozas, E.; Muir, P.; Honkanen, H.; Ilonen, J.; et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 2014, 63, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Andréoletti, L.; Hober, D.; Hober-Vandenberghe, C.; Fajardy, I.; Belaich, S.; Lambert, V.; Vantyghem, M.C.; Lefebvre, J.; Wattre, P. Coxsackie B virus infection and beta cell autoantibodies in newly diagnosed IDDM adult patients. Clin. Diagn. Virol. 1998, 9, 125–133. [Google Scholar] [CrossRef]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-J.; Gim, J.-A.; Lee, J.K.; Park, H.; Shin, O.S. Coxsackievirus B3 Infection of Human Neural Progenitor Cells Results in Distinct Expression Patterns of Innate Immune Genes. Viruses 2020, 12, 325. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.M.; Giannini, R.; Piaggi, P.; Ugolini, C.; Materazzi, G.; Miccoli, P.; Vitti, P.; Basolo, F. A six-gene panel to label follicular adenoma, low- and high-risk follicular thyroid carcinoma. Endocr. Connect. 2018, 7, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.M.; Condello, V.; Denaro, M.; Torregrossa, L.; Elisei, R.; Vitti, P.; Basolo, F. DICER1 somatic mutations strongly impair miRNA processing even in benign thyroid lesions. Oncotarget 2019, 10, 1785–1797. [Google Scholar] [CrossRef]
- Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. [Google Scholar] [CrossRef]
- Toniolo, A.; Leslie, R.D. Diabetes, the TYK2 Gene and the Interferon Response: In Search for Environmental Causes. EBioMedicine 2017, 24, 18–19. [Google Scholar] [CrossRef]
- ICTV Master Species List 2018b.v2. Available online: https://talk.ictvonline.org/files/master-species-lists/m/msl/8266 (accessed on 25 June 2020).
- Stavrou, S.; Ghadge, G.; Roos, R.P. Apoptotic and antiapoptotic activity of L protein of Theiler’s murine encephalomyelitis virus. J. Virol. 2011, 85, 7177–7185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stavrou, S.; Feng, Z.; Lemon, S.M.; Roos, R.P. Different strains of Theiler’s murine encephalomyelitis virus antagonize different sites in the type I interferon pathway. J. Virol. 2010, 84, 9181–9189. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Su, J.; Wang, H.; Chang, J.; Wang, S.; Zheng, W.; Cai, Y.; Wei, W.; Gordy, J.T.; Markham, R.; et al. Disruption of MDA5-Mediated Innate Immune Responses by the 3C Proteins of Coxsackievirus A16, Coxsackievirus A6, and Enterovirus D68. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Nyalwidhe, J.O.; Jurczyk, A.; Satish, B.; Redick, S.; Qaisar, N.; Trombly, M.I.; Vangala, P.; Racicot, R.; Bortell, R.; Harlan, D.M.; et al. Proteomic and Transcriptional Profiles of Human Stem Cell-Derived β Cells Following Enteroviral Challenge. Microorganisms 2020, 8, 295. [Google Scholar] [CrossRef] [PubMed]
- Teo, F.M.S.; Nyo, M.; Wong, A.A.; Tan, N.W.H.; Koh, M.T.; Chan, Y.F.; Chong, C.Y.; Chu, J.J.H. Cytokine and Chemokine Profiling in Patients with Hand, Foot and Mouth Disease in Singapore and Malaysia. Sci. Rep. 2018, 8, 4087. [Google Scholar] [CrossRef]
- Rajendran, S.; Anquetil, F.; Quesada-Masachs, E.; Graef, M.; Gonzalez, N.; McArdle, S.; Chu, T.; Krogvold, L.; Dahl-Jørgensen, K.; von Herrath, M. IL-6 is present in beta and alpha cells in human pancreatic islets: Expression is reduced in subjects with type 1 diabetes. Clin. Immunol. 2020, 211, 108320. [Google Scholar] [CrossRef]
- Dean, J.W.; Peters, L.D.; Fuhrman, C.A.; Seay, H.R.; Posgai, A.L.; Stimpson, S.E.; Brusko, M.A.; Perry, D.J.; Yeh, W.-I.; Newby, B.N.; et al. Innate inflammation drives NK cell activation to impair Treg activity. J. Autoimmun. 2020, 108, 102417. [Google Scholar] [CrossRef] [PubMed]
- Kallionpää, H.; Somani, J.; Tuomela, S.; Ullah, U.; de Albuquerque, R.; Lönnberg, T.; Komsi, E.; Siljander, H.; Honkanen, J.; Härkönen, T.; et al. Early Detection of Peripheral Blood Cell Signature in Children Developing β-Cell Autoimmunity at a Young Age. Diabetes 2019, 68, 2024–2034. [Google Scholar] [CrossRef]
- Thorsen, S.U.; Eising, S.; Mortensen, H.B.; Skogstrand, K.; Pociot, F.; Johannesen, J.; Svensson, J. Danish Childhood Diabetes Registry Systemic levels of CCL2, CCL3, CCL4 and CXCL8 differ according to age, time period and season among children newly diagnosed with type 1 diabetes and their healthy siblings. Scand. J. Immunol. 2014, 80, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More than Just Attractive: How CCL2 Influences Myeloid Cell Behavior beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nPOD(miao) Case No. | Gender | Age (miao) (Years) | Clinical Diagnosis | Diabetes Duration (Years) | C-Peptide (ng/mL) | Virus |
|---|---|---|---|---|---|---|
| Enterovirus-negative | ||||||
| 6385 | M | 10.9 | no diabetes | NA 3 | 1.55 | no |
| 6407 | F | 4.6 | no diabetes | NA 3 | 5.35 | no |
| 6412 | F | 17.6 | no diabetes | NA 3 | 5.60 | no |
| 6420 | M | 11.5 | no diabetes | NA 3 | 1.27 | no |
| Enterovirus-positive | ||||||
| 6063 | M | 4.4 | T1D 1 | 3.0 | 0.04 | Enterovirus |
| 6113 | F | 13.1 | T1D 1 | 1.6 | 0.04 | Enterovirus |
| 6128 | F | 33.8 | T1D 1 | 31.5 | 0.04 | Enterovirus |
| 6181 | M | 31.9 | AAb-pos 2 | NA 3 | 0.06 | Enterovirus |
| Gene | Gene Function | CBV3 vs. CBV3-Ctrl | Spl-EV vs. Spl-Ctrl |
|---|---|---|---|
| Up or Down Regulation | Up or Down Regulation | ||
| Type I IFN Induction Pathway (Response to Viral RNA) | |||
| IFIH1 (MDA5) | Cytoplasmic sensor of viral nucleic acids. Major role in sensing viral infection and in the activating the cascade of antiviral responses including the induction of type I IFNs and proinflammatory cytokines. | - | - |
| TLR3 | Key component of innate and adaptive immunity, a nucleotide-sensing TLR activated by dsRNA. Acts via the adapter TICAM1. | - | - |
| TICAM1 | Component of a multi-helicase-TICAM1 complex acting as a cytoplasmic sensor of viral dsRNA. Activates a cascade of antiviral responses, including proinflammatory cytokines. | - | ↓ |
| TBK1 | Following the activation of toll-like receptors by viral or bacterial components, associates with TRAF3 and TANK and phosphorylates the IFN regulatory factors IRF3 and IRF7 and DDX3X. This activity allows the subsequent nuclear translocation of IRFs leading to the transcriptional activation of type I IFNs and pro-inflammatory cytokines. Activates IRF3 by phosphorylating innate adapters MAVS, TMEM173/STING, TICAM1 thus leading to the recruitment of IRF3. | - | - |
| IFI16 | After binding to viral DNA in the cytoplasm, recruits TMEM173/STING and mediates the induction of IFN-beta. Has anti-inflammatory activity and inhibits the activation of the AIM2 inflammasome. | - | ↓ |
| TMEM173 (STING1) | Facilitator of innate immune signaling that acts as a sensor of cytosolic DNA from viruses and bacteria and promotes the production of type I IFN. | - | ↓ |
| IRAK1 | IL1 receptor-associated kinase 1; phosphorylates interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in the transcriptional activation of type I IFN genes. | - | ↓ |
| TRAF1 | Adapter molecule that regulates the activation of NFKB and JNK. | - | - |
| TRAF2 | Regulates the activation of NFKB and JNK. Regulates cell survival and apoptosis. | - | ↓ |
| TRAF3 | Regulates pathways leading to the activation of NFKB and MAP kinases. Regulates B-cell survival. Part of the signaling pathways leading to the production of IFN and cytokines. Role in T-cell dependent on immune responses. | ↑ | - |
| TRAF4 | Activation of NFKB and JNK in response to signaling through TLRs. Regulates cell survival and apoptosis. | - | ↓ |
| TRAF5 | Mediates the activation of NFKB. | - | ↓ |
| TRAF6 | Activation of NFKB and JUN. Role in dendritic cell maturation and/or activation. | - | ↓ |
| IKBKA (IKKA) | Ikappa kinase (IKK) is an enzyme complex that is part of the NFKB signaling pathway. The IKK complex is comprised of three subunits: alpha, beta, and gamma. The alpha and beta subunits are catalytically active whereas the gamma subunit has regulatory functions. | ↑ | - |
| IKBKB (IKKB) | ↓ | ↓ | |
| IKBKG (NEMO) | - | ↓ | |
| IKBKE (IKKE) | Noncanonical IKB kinase (IKK) that is essential for regulating antiviral signaling pathways. | - | - |
| NFKB1 | NFKB is a homo- or heterodimeric complex formed by the Rel-like domain-containing proteins RELA/p65, RELB, NFKB1/p105, NFKB1/p50, REL, and NFKB2/p52 and the heterodimeric p65-p50 complex. Dimers bind at kappa-B sites in the DNA of target genes and individual dimers have distinct preferences for different kappa-B sites. | ↑ | ↑ |
| NFKB2 | NFKB2 has dual functions such as the cytoplasmic retention of attached NFKB proteins by p100 and the generation of p52 by a cotranslational processing. | - | ↓ |
| NFKBIA | Member of the NFKB inhibitor family. The protein interacts with REL dimers to inhibit NFKB/REL complexes which are involved in inflammatory responses. | - | ↓ |
| NFKBIZ | Member of the ankyrin-repeat family of proteins known to play a role in inflammatory responses. Activates IL6 but decreases TNF-alpha production. | - | ↓ |
| RELA | The NF-kappa-B heterodimeric RELA-NFKB1 and RELA-REL complexes function as transcriptional activators. The NFKB homodimeric RELA-RELA complex activates IL-8 expression. | ↑ | - |
| RELB | The NFKB heterodimeric RelB-p50 and RelB-p52 complexes are transcriptional activators. RELB is required for both T and B lymphocyte maturation and function. | - | - |
| IRF3 | Key transcriptional regulator of type I IFN-dependent immune responses against viruses. Regulates the transcription of type I IFN genes and IFN-stimulated genes by binding to an IFN-stimulated response element (ISRE). | - | ↓ |
| IFITM1 | IFN-induced antiviral protein which inhibits the entry of viruses to the cytoplasm, permitting endocytosis, but preventing subsequent viral fusion and the release of viral contents into the cytosol. | - | ↓ |
| Type I IFN signaling pathway (response to IFN) | |||
| IFNAR1 | Heterodimer with IFNAR2. Type I IFN binding activates the JAK-STAT signaling cascade and triggers the tyrosine phosphorylation of proteins including JAKs, TYK2, STAT, and the IFNR alpha- and beta-subunits themselves. | - | ↓ |
| IFNAR2 | Associates with IFNAR1 to form the type I IFN receptor. Involved in IFN-mediated STAT1, STAT2, and STAT3 activation. Isoforms 1 and 2 are involved in signal transduction due to their association with JAK1. | ↑ | - |
| JAK1 | Tyrosine kinase of the non-receptor type, involved in the IFN-alpha/beta/gamma signal pathway. | ↑ | ↑ |
| JAK2 | Tyrosine kinase of the non-receptor type involved in different processes (cell growth, differentiation, histone modifications). Mediates signaling events in both innate and adaptive immunity. In the cytoplasm, mediates signal transduction via association with type II receptors (IFN-alpha, IFN-beta, IFN-gamma, and multiple interleukins). | - | ↓ |
| TYK2 | Tyrosine kinase; associates with the cytoplasmic domain of type I and type II cytokine receptors and promulgates cytokine signals by phosphorylating receptor subunits. Component of type I and type III IFN signaling pathways. | - | ↓ |
| STAT1 | Transcription activator that mediates cellular responses to IFNs, cytokines, and growth factors. Following type I IFN binding to cell receptors, signaling via protein kinases leads to the activation of TYK2 and JAK1 and to the tyrosine phosphorylation of STAT1 and STAT2. Phosphorylated STATs associate with ISGF3G/IRF-9 the ISGF3 complex transcription factor that enters the nucleus and promotes the transcription of IFN-stimulated genes which drive the cell in an antiviral state. | ↑ | ↑ |
| STAT2 | Signal transducer and activator of transcription that mediates signaling by type I IFNs. | - | ↓ |
| Group Comparison | Pathway | Statistical Mean 1 | q-Value |
|---|---|---|---|
| CBV3 vs. CBV3-Ctrl | hsa04630 JAK-STAT signaling pathway | 1.33 | 0.2741 |
| Spl-EV vs. Spl-Ctrl | hsa04662 B cell receptor signaling pathway | 1.01 | 0.1844 |
| hsa04660 T cell receptor signaling pathway | 0.96 | 0.1844 | |
| hsa04810 Regulation of actin cytoskeleton | 0.97 | 0.1844 | |
| hsa04722 Neurotrophin signaling pathway | 0.95 | 0.1844 | |
| Spl-Ctrl vs. CBV3-Ctrl | hsa04610 Complement and coagulation cascades | 2.68 | <0.0001 |
| Spl-EV vs. CBV3 | hsa04610 Complement and coagulation cascades | 2.65 | <0.0001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poma, A.M.; Genoni, A.; Broccolo, F.; Denaro, M.; Pugliese, A.; Basolo, F.; Toniolo, A. Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes. Microorganisms 2020, 8, 1031. https://doi.org/10.3390/microorganisms8071031
Poma AM, Genoni A, Broccolo F, Denaro M, Pugliese A, Basolo F, Toniolo A. Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes. Microorganisms. 2020; 8(7):1031. https://doi.org/10.3390/microorganisms8071031
Chicago/Turabian StylePoma, Anello Marcello, Angelo Genoni, Francesco Broccolo, Maria Denaro, Alberto Pugliese, Fulvio Basolo, and Antonio Toniolo. 2020. "Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes" Microorganisms 8, no. 7: 1031. https://doi.org/10.3390/microorganisms8071031
APA StylePoma, A. M., Genoni, A., Broccolo, F., Denaro, M., Pugliese, A., Basolo, F., & Toniolo, A. (2020). Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes. Microorganisms, 8(7), 1031. https://doi.org/10.3390/microorganisms8071031