Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Characteristics of Enrolled Patients
2.3. Sample Collection, Processing, DNA Extraction and Sequencing
2.4. Basic Sequence Analyses
2.5. Taxonomic Classification of Metagenomic Contigs
2.6. Bioinformatic and Statistical Analyses
3. Results
3.1. Population and Sampling
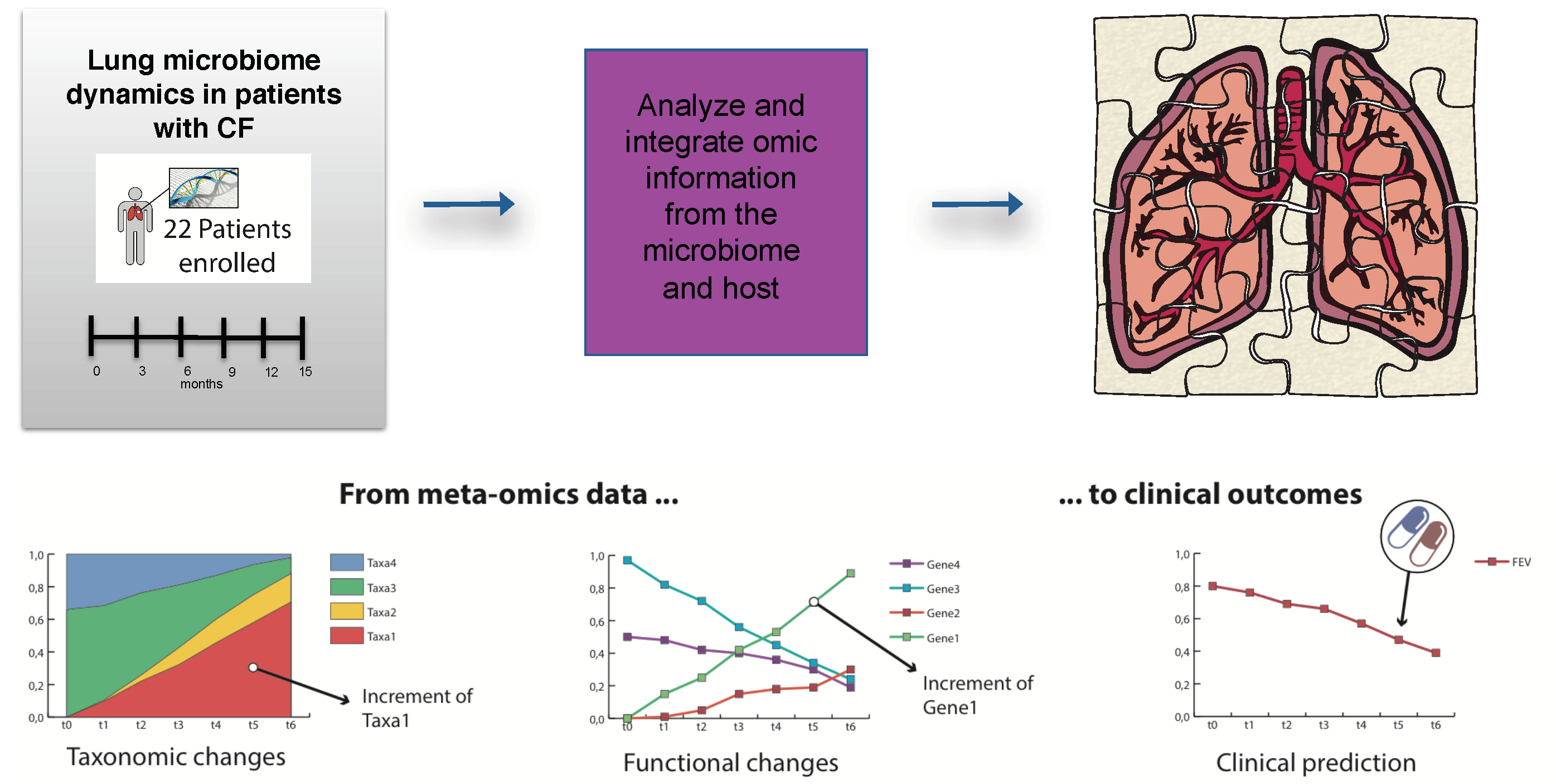
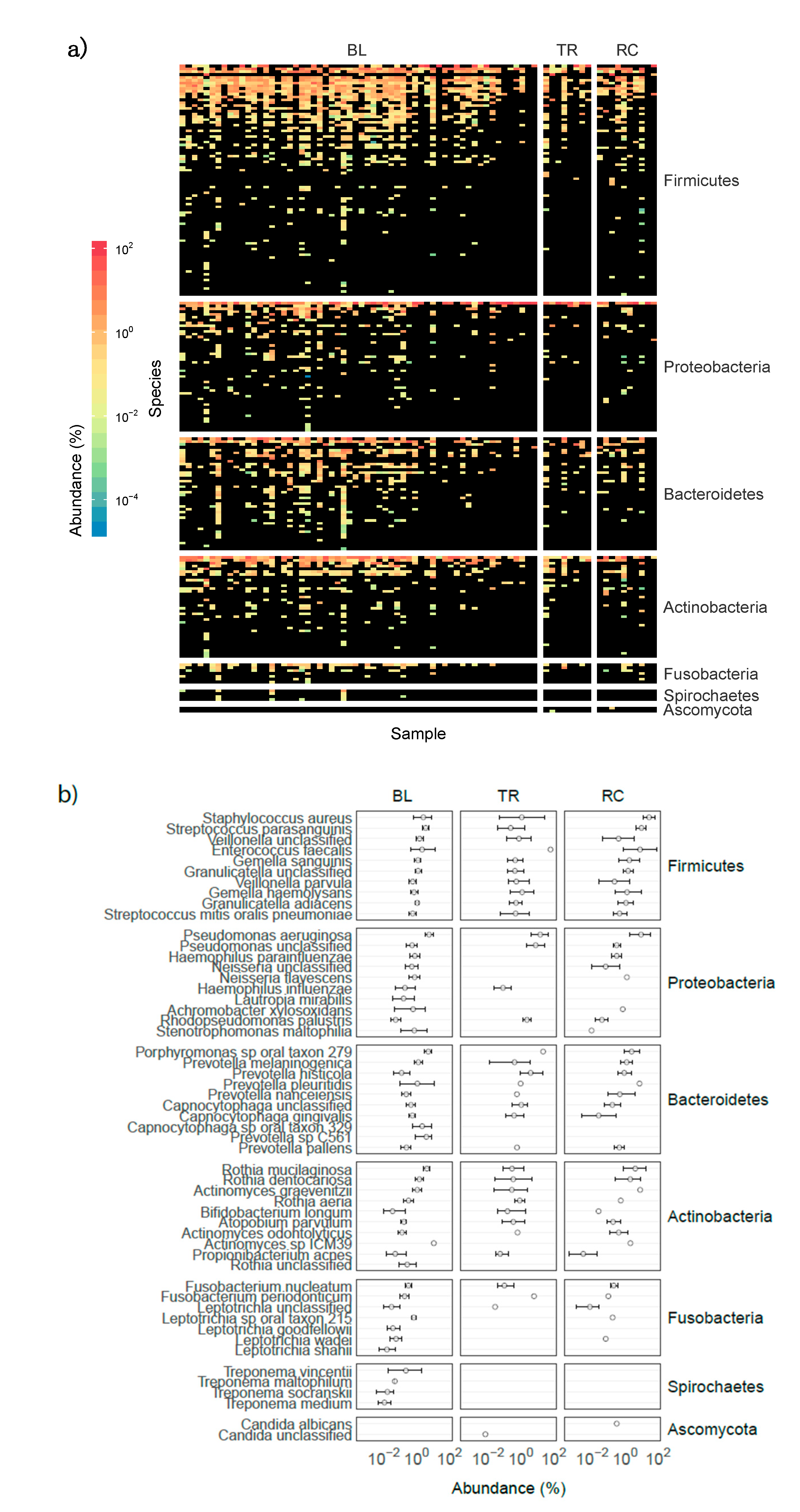
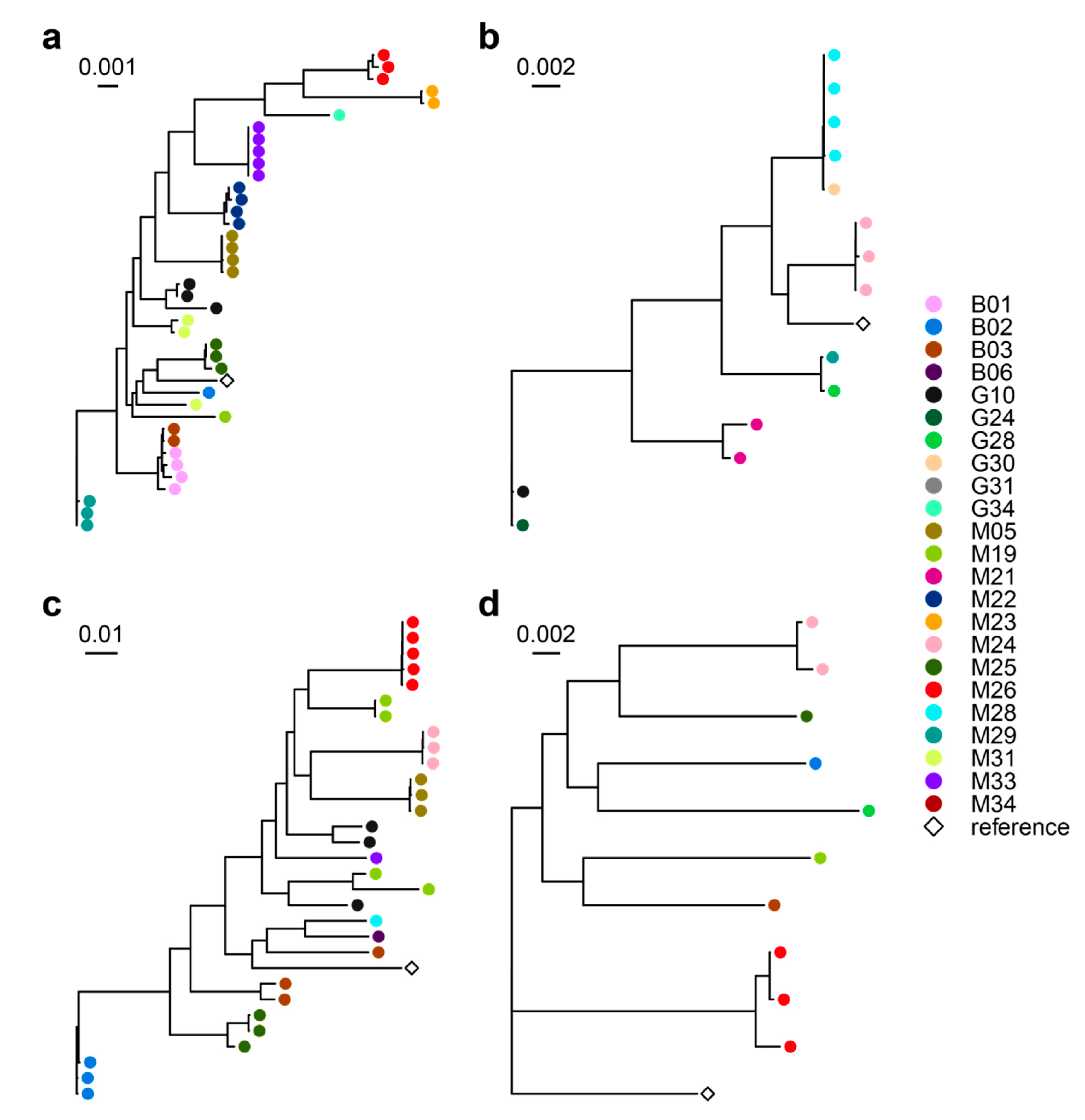
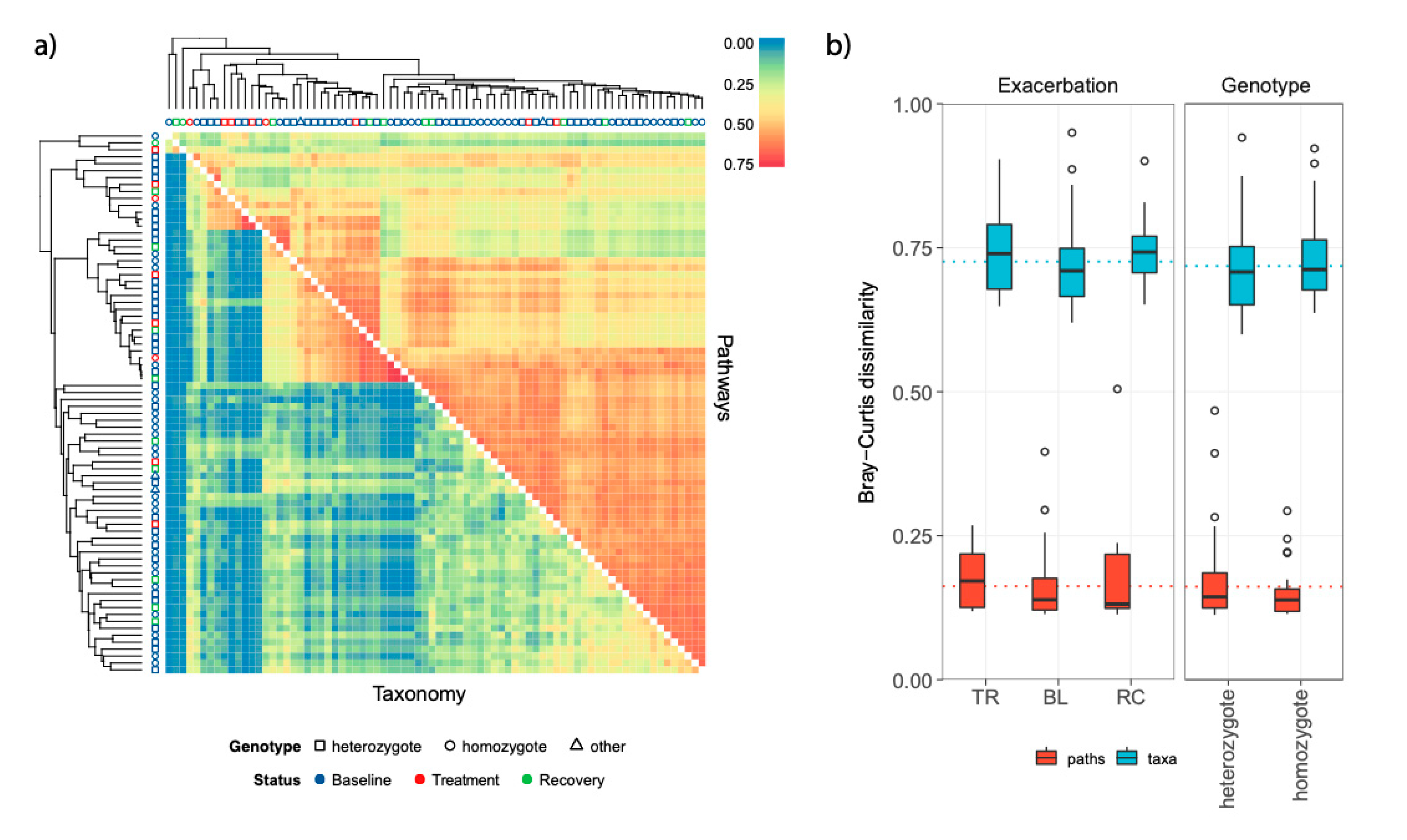
3.2. Airway Microbiomes are Taxonomically Distinct and Show Patient-Specific Strain Colonization
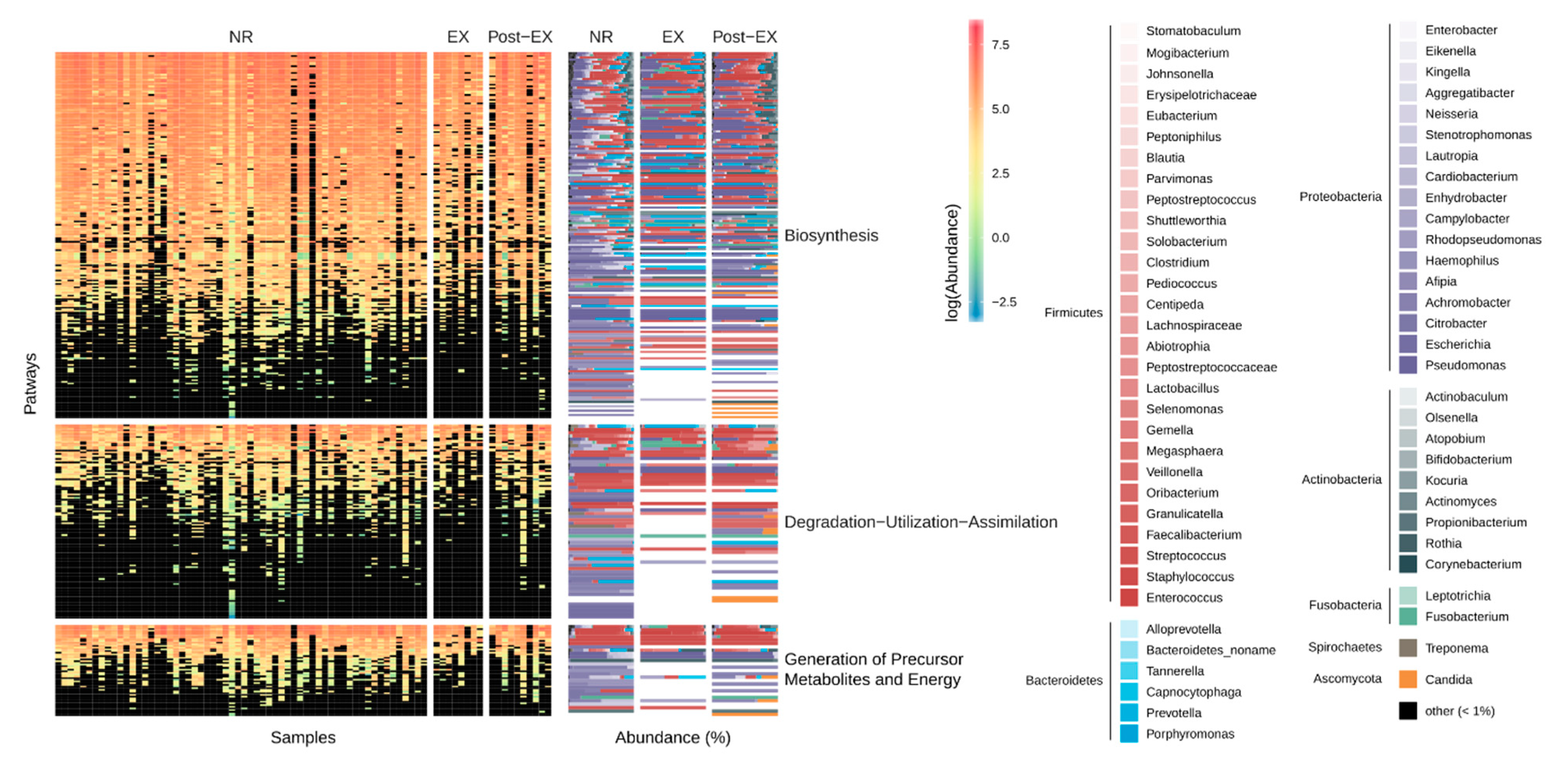
3.3. Stability and Subject-Specific Distribution Patterns of Metagenomic Functions
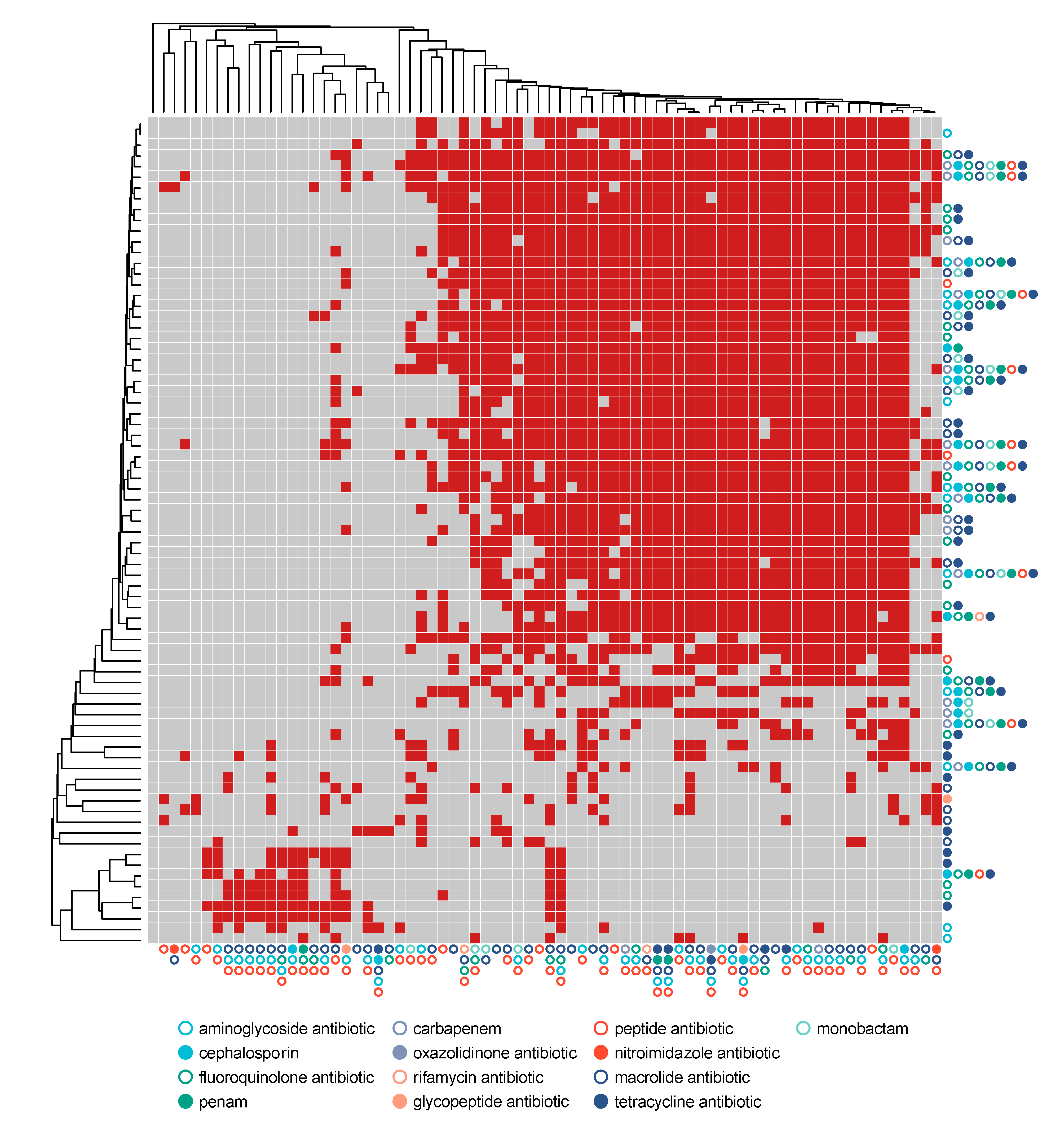
3.4. Resistome Composition through Exacerbation Events and Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Material
References
- Knapp, E.A.; Fink, A.K.; Goss, C.H.; Sewall, A.; Ostrenga, J.; Dowd, C.; Elbert, A.; Petren, K.M.; Marshall, B.C. The Cystic Fibrosis Foundation Patient Registry. Design and methods of a national observational disease registry. Ann. Am. Thorac. Soc. 2016, 13, 1014–1015. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; De Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosisl. Microbiome 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bevivino, A.; Bacci, G.; Drevinek, P.; Nelson, M.T.; Hoffman, L.; Mengoni, A. Deciphering the Ecology of Cystic Fibrosis Bacterial Communities: Towards Systems-Level Integration. Trends Mol. Med. 2019, 25, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Bacci, G.; Mengoni, A.; Fiscarelli, E.; Segata, N.; Taccetti, G.; Dolce, D.; Paganin, P.; Morelli, P.; Tuccio, V.; De Alessandri, A.; et al. A different microbiome gene repertoire in the airways of cystic fibrosis patients with severe lung disease. Int. J. Mol. Sci. 2017, 18, 1654. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef]
- Willner, D.; Daly, J.; Whiley, D.; Grimwood, K.; Wainwright, C.E.; Hugenholtz, P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS ONE 2012, 7, e34605. [Google Scholar] [CrossRef]
- Lim, Y.W.; Evangelista, J.S.; Schmieder, R.; Bailey, B.; Haynes, M.; Furlan, M.; Maughan, H..; Edwards, R.; Rohwer, F.; Conradet, D.; et al. Clinical insights from metagenomic analysis of sputum samples from patients with cystic fibrosis. J. Clin. Microbiol. 2014, 52, 425–437. [Google Scholar] [CrossRef]
- Willner, D.; Haynes, M.R.; Furlan, M.; Hanson, N.; Kirby, B.; Lim, Y.W.; Rainey, P.B.; Schmieder, R.; Youle, M.; Conrad, D.; et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. Am. J. Respir Cell Mol. Biol. 2012, 46, 127–131. [Google Scholar] [CrossRef]
- Whelan, F.J.; Heirali, A.A.; Rossi, L.; Rabin, H.R.; Parkins, M.D.; Surette, M.G. Longitudinal sampling of the lung microbiota in individuals with cystic fibrosis. PLoS ONE 2017, 12, e0172811. [Google Scholar] [CrossRef]
- Whiteson, K.L.; Meinardi, S.; Lim, Y.W.; Schmieder, R.; Maughan, H.; Quinn, R.; Blake, D.R.; Conrad, D.; Rohwer, F. Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. ISME J. 2014, 8, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Graeber, S.Y.; Stahl, M.; Dittrich, A.S.; Mall, M.A.; Dalpke, A.H. Chronic but not intermittent infection with Pseudomonas aeruginosa is associated with global changes of the lung microbiome in cystic fibrosis. Eur. Respir. J. 2017, 50, 1701086. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Ward, D.V.; Pasolli, E.; Tolio, T.; Zolfo, M.; Asnicar, F.; Truong, D.T.; Tett, A.; Morrow, A.L.; Segata, N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 2016, 13, 435–438. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef]
- Proesmans, M.; Balinska-Miskiewicz, W.; Dupont, L.; Bossuyt, X.; Verhaegen, J.; Høiby, N.; de Boeck, K. Evaluating the ‘Leeds criteria’ for Pseudomonas aeruginosa infection in a cystic fibrosis centre. Eur. Respir. J. 2006, 27, 937–943. [Google Scholar] [CrossRef]
- Bacci, G.; Paganin, P.; Lopez, L.; Vanni, C.; Dalmastri, C.; Cantale, C.; Daddiego, L.; Perrotta, G.; Dolce, D.; Morelli, P.; et al. Pyrosequencing Unveils Cystic Fibrosis Lung Microbiome Differences Associated with a Severe Lung Function Decline. PLoS ONE 2016, 11, e0156807. [Google Scholar]
- Flume, P.A.; Mogayzel, P.J.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef]
- Taylor-Robinson, D.; Whitehead, M.; Diderichsen, F.; Olesen, H.V.; Pressler, T.; Smyth, R.L.; Diggle, P. Understanding the natural progression in %FEV1 decline in patients with cystic fibrosis: A longitudinal study. Thorax 2012, 67, 860–866. [Google Scholar] [CrossRef]
- Kerem, E.; Conway, S.; Elborn, S.; Heijerman, H.; Consensus Committee. Standards of care for patients with cystic fibrosis: A European consensus. J. Cyst, Fibros 2005, 4, 7–26. [Google Scholar] [CrossRef]
- Paganin, P.; Fiscarelli, E.V.; Tuccio, V.; Chiancianesi, M.; Bacci, G.; Morelli, P.; Dolce, D.; Dalmastri, C.; De Alessandri, A.; Lucidi, V.; et al. Changes in Cystic Fibrosis Airway Microbial Community Associated with a Severe Decline in Lung Function. PLoS ONE 2015, 10, e0124348. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; Rosenstein, B.J.; White, T.B.; Accurso, F.J.; Castellani, C.; Cutting, G.R.; Durie, P.R.; Legrys, V.A.; Massie, J.; Parad, R.B.; et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J. Pediatr. 2008, 153, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Bazzicalupo, M.; Benedetti, A.; Mengoni, A. StreamingTrim 1.0: A Java software for dynamic trimming of 16S rRNA sequence data from metagenetic studies. Mol. Ecol. Resour. 2014, 14, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Abubucker, S.; Segata, N.; Goll, J.; Schubert, A.M.; Izard, J.; Cantarel, B.L.; Rodriguez-Mueller, B.; Zucker, J.; Thiagarajan, M.; Henrissat, B.; et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012, 8, e1002358. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segatal, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Truong, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012; Available online: http://www.R-project.org/ (accessed on 25 May 2020).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Faust, K.; Lahti, L.; Gonze, D.; de Vos, W.M.; Raes, J. Metagenomics meets time series analysis: Unraveling microbial community dynamics. Curr. Opin. Microbiol. 2015, 25, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of Microbial Community Resistance and Resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Segata, N. On the Road to Strain-Resolved Comparative Metagenomics. mSystems 2018, 3. [Google Scholar] [CrossRef]
- Zolfo, M.; Asnicar, F.; Manghi, P.; Pasolli, E.; Tett, A.; Segata, N. Profiling microbial strains in urban environments using metagenomic sequencing data. Biol. Direct. 2018, 13, 1–13. [Google Scholar] [CrossRef]
- Carmody, L.A.; Zhao, J.; Kalikin, L.M.; LeBar, W.; Simon, R.H.; Venkataraman, A.; Schmidt, T.M.; Abdo, Z.; Schloss, P.D.; LiPuma, J.J. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 2015, 3, 12. [Google Scholar] [CrossRef]
- Cox, M.J.; Turek, E.M.; Hennessy, C.; Mirza, G.K.; James, P.L.; Coleman, M.; Jones, A.; Wilson, R.; Bilton, D.; Cookson, W.O.C.; et al. Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS ONE 2017, 12, e0170622. [Google Scholar] [CrossRef]
- Fodor, A.A.; Klem, E.R.; Gilpin, D.F.; Elborn, J.S.; Boucher, R.C.; Tunney, M.M.; Wolfgang, M.C. The Adult Cystic Fibrosis Airway Microbiota Is Stable over Time and Infection Type, and Highly Resilient to Antibiotic Treatment of Exacerbations. PLoS ONE 2012, 7, e45001. [Google Scholar] [CrossRef] [PubMed]
- Carmody, L.A.; Zhao, J.; Schloss, P.D.; Petrosino, J.F.; Murray, S.; Young, V.B.; Li, J.Z.; LiPuma, J.J. Changes in Cystic Fibrosis Airway Microbiota at Pulmonary Exacerbation. Ann. Am. Thorac. Soc. 2013, 10, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Zemanick, E.T.; Wagner, B.D.; Robertson, C.E.; Ahrens, R.C.; Chmiel, J.F.; Clancy, J.P.; Gibson, R.L.; Harris, W.T.; Kurland, G.; Laguna, T.A.; et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur. Respir. J. 2017, 50, 1700832. [Google Scholar] [CrossRef] [PubMed]
- Carmody, L.A.; Caverly, L.J.; Foster, B.K.; Rogers, M.A.M.; Kalikin, L.M.; Simon, R.H.; VanDevanter, D.R.; LiPuma, J.J. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS ONE 2018, 13, e0194060. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F.; Booth, A. It’s the song, not the singer: An exploration of holobiosis and evolutionary theory. Biol. Philos. 2016, 32, 5–24. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Itav, S.; Rothschild, D.; Meijer, M.; Levy, M.; Moresi, C.; Dohnalová, L.; Braverman, S.; Rozin, S.; Malitsky, S.; et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 2016, 540, 1–27. [Google Scholar] [CrossRef]
- Allemann, A.; Kraemer, J.G.; Korten, I.; Ramsey, K.; Casaulta, C.; Wüthrich, D.; Ramette, A.; Endimiani, A.; Latzin, P.; Hilty, M. Nasal resistome development in infants with cystic fibrosis in the first year of life. Front. Microbiol. 2019, 10, 212. [Google Scholar] [CrossRef]
- Feigelman, R.; Kahlert, C.R.; Baty, F.; Rassouli, F.; Kleiner, R.L.; Kohler, P.; Brutsche, M.H.; von Mering, C. Sputum DNA sequencing in cystic fibrosis: Non-invasive access to the lung microbiome and to pathogen details. Microbiome 2017, 5, 20. [Google Scholar] [CrossRef]
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The versatile mutational resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 685. [Google Scholar] [CrossRef]
- López-Causapé, C.; Oliver, A. Insights into the evolution of the mutational resistome of Pseudomonas aeruginosa in cystic fibrosis. Future Microbiol. 2017, 12, 1445–1448. [Google Scholar] [CrossRef]
- Taylor, S.L.; Leong, L.E.X.; Mobegi, F.M.; Choo, J.M.; Burr, L.D.; Wesselingh, S.; Rogers, G.B. Understanding the impact of antibiotic therapies on the respiratory tract resistome: A novel pooled-template metagenomic sequencing strategy. Multidiscip. Respir. Med. 2018, 13, 30. [Google Scholar] [CrossRef]
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes (Basel) 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Genotype | Gender | FEV1 Status | Age | n | EX | %FEV1 |
|---|---|---|---|---|---|---|---|
| B01 | ΔF508/2183AA->G | M | S | 27 | 5 | yes | 37.0 ± 1.70 |
| B02 | ΔF508/N1303K | F | SD | 26 | 3 | no | 54.7 ± 3.48 |
| B03 | ΔF508/4016insT | F | S | 30 | 4 | no | 55.0 ± 1.08 |
| B06 | ΔF508/ΔF508 | F | SD | 21 | 4 | no | 60.2 ± 3.42 |
| G10 | ΔF508/ΔF508 | M | S | 51 | 4 | no | 54.0 ± 3.08 |
| G24 | ΔF508/ΔF508 | F | S | 49 | 3 | yes | 31.0 ± 4.08 |
| G28 | ΔF508/ΔF508 | F | NA | 38 | 2 | no | 42.5 ± 1.50 |
| G30 | ΔF508/ΔF508 | F | S | 50 | 1 | no | 54 |
| G31 | G1244E/G42X | F | SD | 53 | 2 | no | 41.5 ± 1.50 |
| G34 | ΔF508/ΔF508 | F | S | 39 | 1 | no | 47 |
| M05 | ΔF508/ΔF508 | M | SD | 32 | 4 | no | 34.8 ± 0.85 |
| M19 | ΔF508/ΔF508 | M | S | 24 | 4 | no | 44.0 ± 2.04 |
| M21 | ΔF508/N1303K | M | SD | 27 | 4 | yes | 51.5 ± 4.35 |
| M22 | ΔF508/2789+5G->A | F | S | 29 | 5 | yes | 50.4 ± 1.03 |
| M23 | ΔF508/G542X | F | S | 30 | 4 | yes | 37.0 ± 1.47 |
| M24 | ΔF508/ΔF508 | M | S | 32 | 3 | no | 35.2 ± 0.85 |
| M25 | ΔF508/296+1G->T | F | SD | 41 | 4 | no | 42.5 ± 2.02 |
| M26 | ΔF508/3849+10 | F | SD | 49 | 5 | yes | 39.6 ± 1.94 |
| M28 | ΔF508/N1303K | M | S | 23 | 4 | no | 39.0 ± 1.08 |
| M29 | ΔF508/G542X | F | S | 12 | 4 | no | 43.5 ± 3.75 |
| M31 | ΔF508/ΔF508 | F | SD | 11 | 3 | yes | 32.7 ± 4.41 |
| M33 | ΔF508/G85E | F | SD | 13 | 5 | yes | 35.4 ± 5.78 |
| Total: 22 | Heterozygote:11 Homozygote:10 Other:1 | F:15 M:7 | S:12 SD:9 | 32.1 ± 2.73 | 78 | no:14 yes:8 | 43.5 ± 1.09 |
| Df | SumOf Sqs | R2 | F | Pr(>F) | |
|---|---|---|---|---|---|
| TAXONOMY | |||||
| Status | 2 | 0.68 | 0.03 | 1.91 | 0.0300 |
| Genotype | 1 | 0.77 | 0.03 | 4.30 | 0.0020 |
| Subject | 18 | 11.97 | 0.52 | 3.74 | 0.0010 |
| FEV1 value | 1 | 0.27 | 0.01 | 1.53 | 0.1349 |
| Days | 1 | 0.28 | 0.01 | 1.58 | 0.1229 |
| Status:Genotype | 1 | 0.11 | 0.01 | 0.64 | 0.7642 |
| Residual | 49 | 8.72 | 0.38 | - | - |
| PATHWAY | |||||
| Status | 2 | 0.20 | 0.04 | 2.37 | 0.0220 |
| Genotype | 1 | 0.14 | 0.03 | 3.42 | 0.0080 |
| Subject | 18 | 2.43 | 0.48 | 3.20 | 0.0010 |
| FEV1 value | 1 | 0.09 | 0.02 | 2.14 | 0.0989 |
| Days | 1 | 0.05 | 0.01 | 1.26 | 0.2458 |
| Status:Genotype | 1 | 0.08 | 0.02 | 1.96 | 0.1169 |
| Residual | 49 | 2.07 | 0.41 | - | - |
| Gene Name | Gene Family | Resistance Mechanism | Drug Class | Antibiotic Class | logFC | AveExpr | t | P.Value | adj.P.Val |
|---|---|---|---|---|---|---|---|---|---|
| basS | pmr phosphoethanolamine transferase | antibiotic target alteration | peptide antibiotic | peptide antibiotic | −0.80 | 11.51 | −5.22 | <0.00001 | 0.0001 |
| FosA | fosfomycin thiol transferase | antibiotic inactivation | Fosfomycin | peptide antibiotic | −1.10 | 10.08 | −3.56 | 0.0006 | 0.0199 |
| ArmR | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux | aminocoumarin antibiotic; carbapenem; cephalosporin; cephamycin; diaminopyrimidine antibiotic; fluoroquinolone antibiotic; macrolide antibiotic; monobactam; penam; penem; peptide antibiotic; phenicol antibiotic; sulfonamide antibiotic; tetracycline antibiotic | peptide antibiotic | −1.56 | 9.24 | −3.42 | 0.0010 | 0.0213 |
| OXA-50 | OXA beta-lactamase | antibiotic inactivation | cephalosporin; penam | aminoglycoside antibiotic | −0.45 | 11.61 | −3.51 | 0.0008 | 0.0397 |
| Pseudomonas aeruginosa soxR | ATP-binding cassette (ABC) antibiotic efflux pump; major facilitator superfamily (MFS) antibiotic efflux pump; resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux; antibiotic target alteration | acridine dye; cephalosporin; fluoroquinolone antibiotic; glycylcycline; penam; phenicol antibiotic; rifamycin antibiotic; tetracycline antibiotic; triclosan | fluoroquinolone antibiotic | −1.28 | 10.13 | −4.43 | <0.00001 | 0.0020 |
| MexR | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux; antibiotic target alteration | aminocoumarin antibiotic; carbapenem; cephalosporin; cephamycin; diaminopyrimidine antibiotic; fluoroquinolone antibiotic; macrolide antibiotic; monobactam; penam; penem; peptide antibiotic; phenicol antibiotic; sulfonamide antibiotic; tetracycline antibiotic | fluoroquinolone antibiotic | −0.73 | 11.11 | −3.30 | 0.0015 | 0.0454 |
| MexR | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux; antibiotic target alteration | aminocoumarin antibiotic; carbapenem; cephalosporin; cephamycin; diaminopyrimidine antibiotic; fluoroquinolone antibiotic; macrolide antibiotic; monobactam; penam; penem; peptide antibiotic; phenicol antibiotic; sulfonamide antibiotic; tetracycline antibiotic | monobactam | 1.75 | 10.90 | 4.95 | <0.00001 | 0.0004 |
| mdtO | major facilitator superfamily (MFS) antibiotic efflux pump | antibiotic efflux | acridine dye; nucleoside antibiotic | monobactam | 2.13 | 10.40 | 3.73 | 0.0004 | 0.0150 |
| OpmD | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux | acridine dye; fluoroquinolone antibiotic; tetracycline antibiotic | monobactam | −2.15 | 10.44 | −3.28 | 0.0016 | 0.0414 |
| MexT | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux | diaminopyrimidine antibiotic; fluoroquinolone antibiotic; phenicol antibiotic | monobactam | −1.52 | 10.57 | −3.19 | 0.0021 | 0.0414 |
| MexK | resistance-nodulation-cell division (RND) antibiotic efflux pump | antibiotic efflux | macrolide antibiotic; tetracycline antibiotic; triclosan | nitroimidazole antibiotic | −3.85 | 11.97 | −3.71 | 0.0004 | 0.0407 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacci, G.; Taccetti, G.; Dolce, D.; Armanini, F.; Segata, N.; Di Cesare, F.; Lucidi, V.; Fiscarelli, E.; Morelli, P.; Casciaro, R.; et al. Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease. Microorganisms 2020, 8, 1003. https://doi.org/10.3390/microorganisms8071003
Bacci G, Taccetti G, Dolce D, Armanini F, Segata N, Di Cesare F, Lucidi V, Fiscarelli E, Morelli P, Casciaro R, et al. Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease. Microorganisms. 2020; 8(7):1003. https://doi.org/10.3390/microorganisms8071003
Chicago/Turabian StyleBacci, Giovanni, Giovanni Taccetti, Daniela Dolce, Federica Armanini, Nicola Segata, Francesca Di Cesare, Vincenzina Lucidi, Ersilia Fiscarelli, Patrizia Morelli, Rosaria Casciaro, and et al. 2020. "Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease" Microorganisms 8, no. 7: 1003. https://doi.org/10.3390/microorganisms8071003
APA StyleBacci, G., Taccetti, G., Dolce, D., Armanini, F., Segata, N., Di Cesare, F., Lucidi, V., Fiscarelli, E., Morelli, P., Casciaro, R., Negroni, A., Mengoni, A., & Bevivino, A. (2020). Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease. Microorganisms, 8(7), 1003. https://doi.org/10.3390/microorganisms8071003