Spatial Variation in Bacterioplankton Communities in the Pearl River, South China: Impacts of Land Use and Physicochemical Factors



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site and Sampling
2.2. Physicochemical Analyses and Land Use Data
2.3. DNA Extraction, PCR Amplification, and 16S rRNA Sequencing
2.4. Processing of Sequencing Data
2.5. Statistical Analysis
3. Results
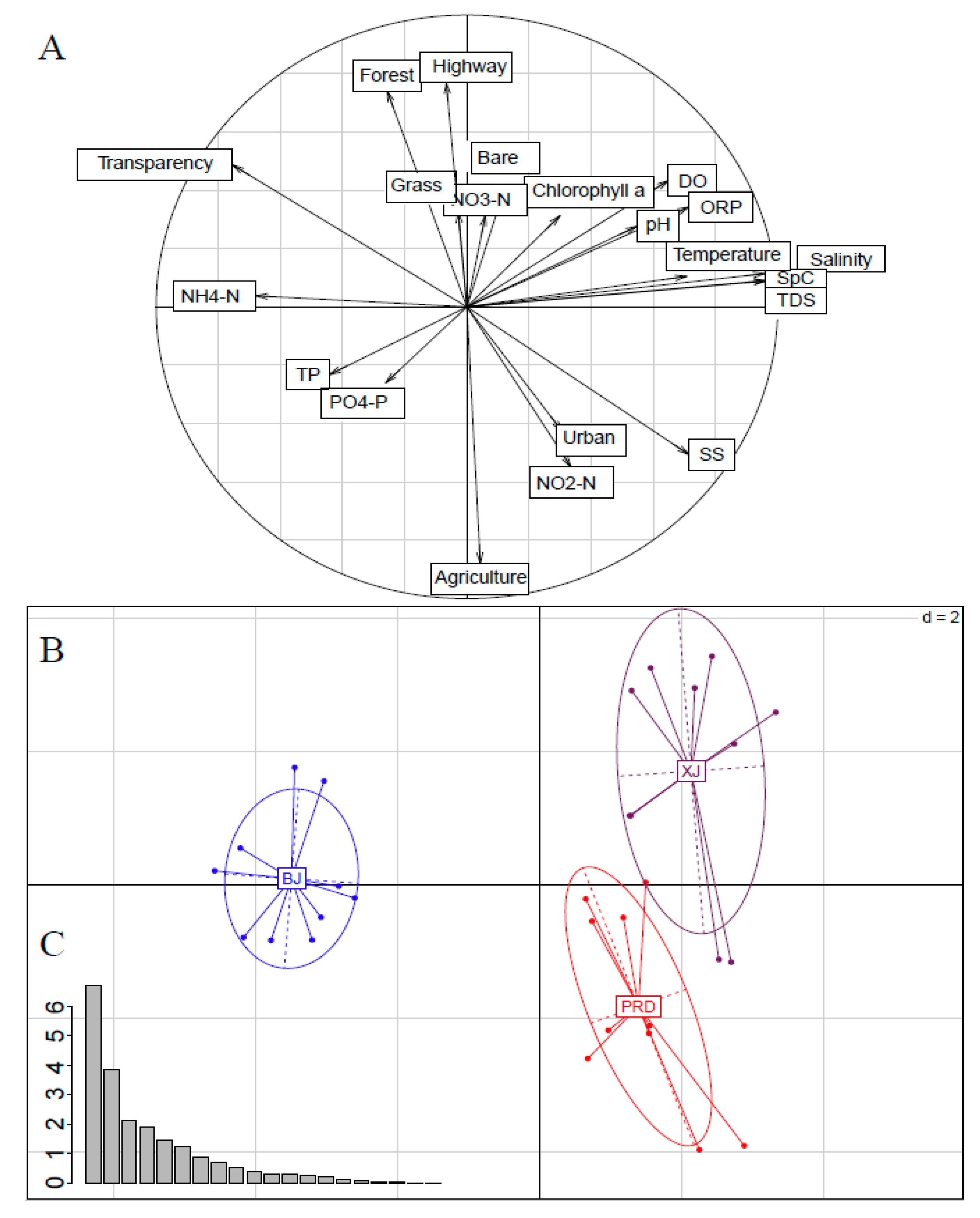
3.1. Environmental Variables
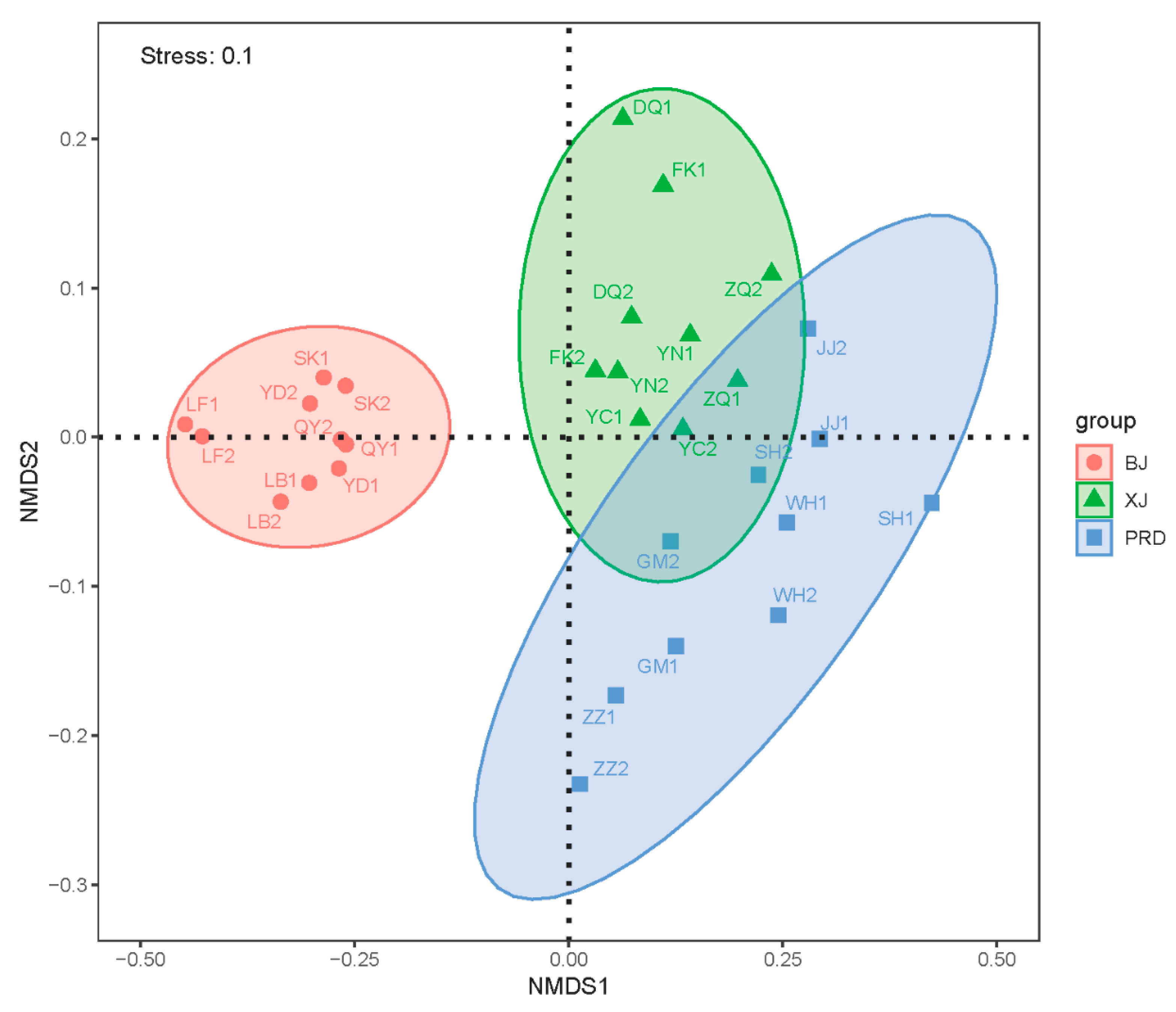
3.2. Bacterioplankton Community Composition, Diversity, and Spatial Variations
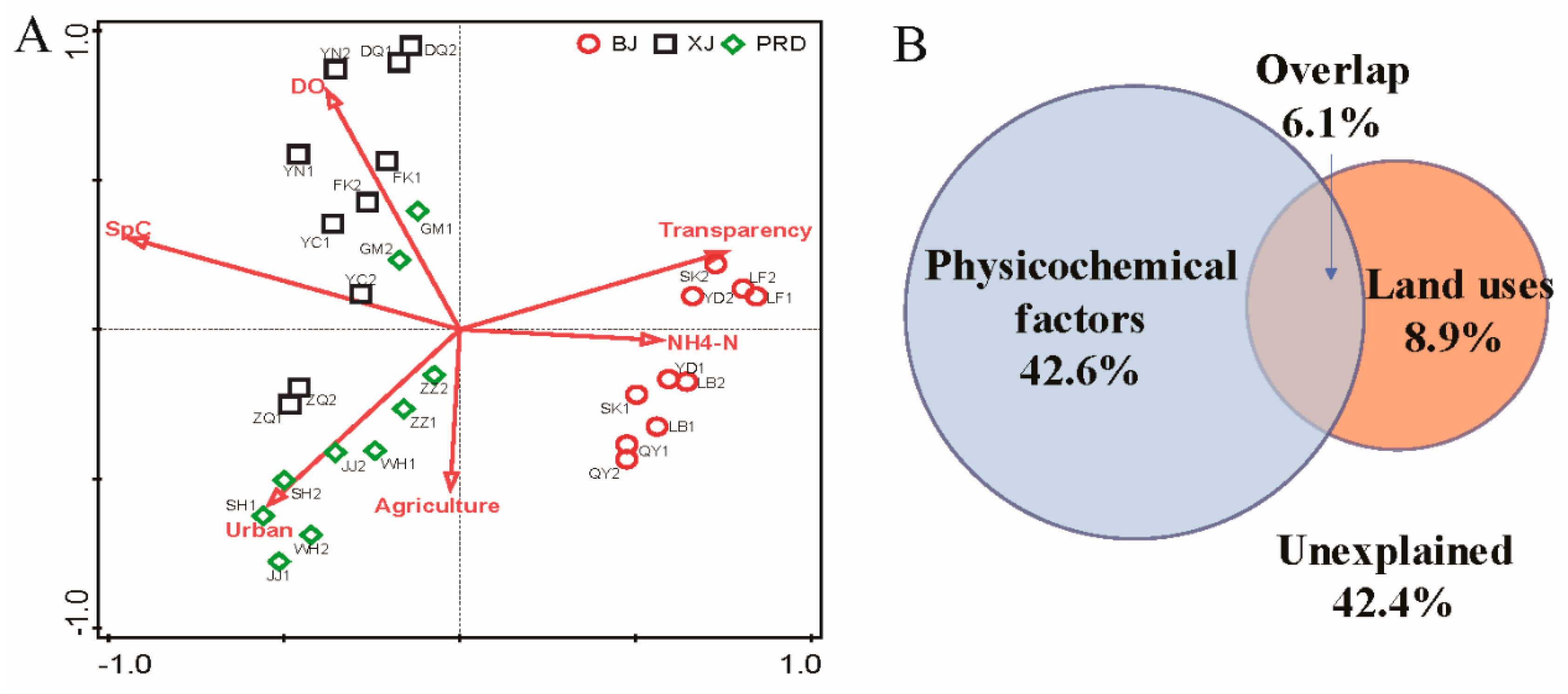
3.3. Effects of Physicochemical Variables and Land Use on Bacterioplankton Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| XJ | Xijiang River |
| BJ | Beijing River |
| PRD | Pearl River Delta |
| DO | Dissolved oxygen |
| TDS | Total dissolved solids |
| SpC | Specific conductivity |
| ORP | Oxidation-reduction potential |
| NH4-N | Ammonium |
| NO3-N | Nitrate |
| NO2-N | Nitrite |
| TP | Total phosphorus |
| PO4-P | Phosphate |
| SS | Suspended solids |
| OTU | Operational taxonomic units |
| PCA | Principal component analysis |
| NMDS | Nonmetric multidimensional scaling |
| ANOSIM | Analysis of similarity |
| LDA | Linear discriminant analysis |
| LEfSe | Linear effect size analysis |
| RDA | Redundancy analysis |
| VPA | Variation partitioning analysis |
| PICRUSt | Phylogenetic investigation of communities by reconstruction of unobserved states |
References
- Azam, F.; Worden, A.Z. Oceanography. Microbes, molecules, and marine ecosystems. Science 2004, 303, 1622–1624. [Google Scholar] [PubMed]
- Nam, Y.D.; Sung, Y.; Chang, H.W.; Roh, S.W.; Kim, K.H.; Rhee, S.K.; Kim, J.C.; Kim, J.Y.; Yoon, J.H.; Bae, J.W. Characterization of the depth-related changes in the microbial communities in Lake Hovsgol sediment by 16S rRNA gene-based approaches. J. Microbiol. 2008, 46, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Xia, X.H.; Liu, T.; Hu, L.J.; Zhu, B.T.; Zhang, X.T.; Dong, J.W. Characteristics of bacterial community in the water and surface sediment of the Yellow River, China, the largest turbid river in the world. J. Soils Sediment. 2014, 14, 1894–1904. [Google Scholar] [CrossRef]
- Chapin, F.S., 3rd; Zavaleta, E.S.; Eviner, V.T.; Naylor, R.L.; Vitousek, P.M.; Reynolds, H.L.; Hooper, D.U.; Lavorel, S.; Sala, O.E.; Hobbie, S.E.; et al. Consequences of changing biodiversity. Nature 2000, 405, 234–242. [Google Scholar] [PubMed]
- Green, J.L.; Bohannan, B.J.; Whitaker, R.J. Microbial biogeography: From taxonomy to traits. Science 2008, 320, 1039–1043. [Google Scholar] [CrossRef]
- Winter, C.; Hein, T.; Kavka, G.; Mach, R.L.; Farnleitner, A.H. Longitudinal changes in the bacterial community composition of the Danube River: A whole-river approach. Appl. Environ. Microbiol. 2007, 73, 421–431. [Google Scholar]
- Ma, L.L.; Mao, G.N.; Liu, J.; Gao, G.H.; Zou, C.L.; Bartlam, M.G.; Wang, Y.Y. Spatial-Temporal Changes of Bacterioplankton Community along an Exhorheic River. Front. Microbiol. 2016, 7, 171. [Google Scholar] [CrossRef]
- Doherty, M.; Yager, P.L.; Moran, M.A.; Coles, V.J.; Fortunato, C.S.; Krusche, A.V.; Medeiros, P.M.; Payet, J.P.; Richey, J.E.; Satinsky, B.M.; et al. Bacterial Biogeography across the Amazon River-Ocean Continuum. Front. Microbiol. 2017, 8, 882. [Google Scholar] [CrossRef]
- Kopylov, A.I.; Ievleva, T.V.; Romanenko, A.V.; Zabotkina, E.A. Structural and Functional Characteristics of the Bacterioplankton of Rivers Flowing through a Large City (Cherepovets, Upper Volga Region). Biol. Bull. 2016, 43, 1350–1356. [Google Scholar] [CrossRef]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Wei, D.; Deng, X.; Weng, S.; He, Z.; He, J. Environmental Factors Shape Water Microbial Community Structure and Function in Shrimp Cultural Enclosure Ecosystems. Front. Microbiol. 2017, 8, 2359. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Liu, L.M.; Chen, H.H.; Yang, J. Spatiotemporal dynamics and determinants of planktonic bacterial and microeukaryotic communities in a Chinese subtropical river. Appl. Microbiol. Biot. 2015, 99, 9255–9266. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.A.; Leff, L.G. Nutrients and other abiotic factors affecting bacterial communities in an Ohio River (USA). Microb. Ecol. 2007, 54, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lei, M.; Li, Y.; Wang, P.; Wang, C.; Gao, Y.; Wu, H.; Xu, C.; Niu, L.; Wang, L.; et al. Determination of vertical and horizontal assemblage drivers of bacterial community in a heavily polluted urban river. Water Res. 2019, 161, 98–107. [Google Scholar] [CrossRef]
- Staley, C.; Gould, T.J.; Wang, P.; Phillips, J.; Cotner, J.B.; Sadowsky, M.J. Bacterial community structure is indicative of chemical inputs in the Upper Mississippi River. Front. Microbiol. 2014, 5, 524. [Google Scholar] [CrossRef]
- Abia, A.L.K.; Alisoltani, A.; Keshri, J.; Ubomba-Jaswa, E. Metagenomic analysis of the bacterial communities and their functional profiles in water and sediments of the Apies River, South Africa, as a function of land use. Sci. Total. Environ. 2018, 616, 326–334. [Google Scholar] [CrossRef]
- Shuai, F.M.; Li, X.H.; Chen, F.C.; Li, Y.F.; Lek, S. Spatial patterns of fish assemblages in the Pearl River, China: Environmental correlates. Fund. Appl. Limnol. 2017, 189, 329–340. [Google Scholar] [CrossRef]
- Zhou, L.; Zeng, L.; Fu, D.H.; Xu, P.; Zeng, S.; Tang, Q.D.; Chen, Q.F.; Chen, L.A.; Li, G.F. Fish density increases from the upper to lower parts of the Pearl River Delta, China, and is influenced by tide, chlorophyll-a, water transparency, and water depth. Aquat. Ecol. 2016, 50, 59–74. [Google Scholar] [CrossRef]
- Peng, X.Z.; Xiong, S.S.; Ou, W.H.; Wang, Z.F.; Tan, J.H.; Jin, J.B.; Tang, C.M.; Liu, J.; Fan, Y.J. Persistence, temporal and spatial profiles of ultraviolet absorbents and phenolic personal care products in riverine and estuarine sediment of the Pearl River catchment, China. J. Hazard. Mater. 2017, 323, 139–146. [Google Scholar] [CrossRef]
- Tan, X.C.; Li, X.H.; Lek, S.; Li, Y.F.; Wang, C.; Li, J.; Luo, J.R. Annual dynamics of the abundance of fish larvae and its relationship with hydrological variation in the Pearl River. Environ. Biol. Fishes 2010, 88, 217–225. [Google Scholar] [CrossRef]
- Zhou, W.H.; Long, A.M.; Jiang, T.; Chen, S.Y.; Huang, L.M.; Huang, H.; Cai, C.H.; Yan, Y. Bacterioplankton dynamics along the gradient from highly eutrophic Pearl River Estuary to oligotrophic northern South China Sea in wet season: Implication for anthropogenic inputs. Mar. Pollut. Bull. 2011, 62, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.W.; Fu, B.B.; Yang, H.M.; Zhao, M.X.; He, B.Y.; Zhang, X.H. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: The potential impact of hypoxia and nutrients. Front. Microbiol. 2015, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Jiang, X.; Jing, Z.Y.; Li, G.; Chen, Z.Z.; Zhou, L.B.; Zhao, C.Y.; Liu, J.X.; Tan, Y.H. Spatial and seasonal distributions of bacterioplankton in the Pearl River Estuary: The combined effects of riverine inputs, temperature, and phytoplankton. Mar. Pollut. Bull. 2017, 125, 199–207. [Google Scholar] [CrossRef]
- Li, J.L.; Salam, N.; Wang, P.D.; Chen, L.X.; Jiao, J.Y.; Li, X.; Xian, W.D.; Han, M.X.; Fang, B.Z.; Mou, X.Z.; et al. Discordance Between Resident and Active Bacterioplankton in Free-Living and Particle-Associated Communities in Estuary Ecosystem. Microb. Ecol. 2018, 76, 637–647. [Google Scholar] [CrossRef]
- Sun, W.; Xia, C.Y.; Xu, M.Y.; Guo, J.; Sun, G.P. Application of modified water quality indices as indicators to assess the spatial and temporal trends of water quality in the Dongjiang River. Ecol. Indic. 2016, 66, 306–312. [Google Scholar] [CrossRef]
- Jin, X.C.; Tu, Q.Y. The Standard Methods for Observation and Analysis of Lake Eutrophication, 2nd ed.; China Environmental Science Press: Beijing, China, 1990. [Google Scholar]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appli. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596, (Database issue). [Google Scholar] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R. Package ‘Vegan’ Community Ecology Package; Version 2.5–6. 2013. Available online: https://rdrr.io/cran/vegan/ (accessed on 1 May 2020).
- Šmilauer, P.; Lepš, J. Multivariate Analysis of Ecological Data Using CANOCO 5[M]; Cambridge University Press: New York, NY, USA, 2014. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Peter, H.; Sommaruga, R. Shifts in diversity and function of lake bacterial communities upon glacier retreat. ISME J. 2016, 10, 1545–1554. [Google Scholar] [CrossRef]
- De’ath, G. Boosted trees for ecological modeling and prediction. Ecology 2007, 88, 243–251. [Google Scholar]
- Crump, B.C.; Peterson, B.J.; Raymond, P.A.; Amon, R.M.W.; Rinehart, A.; McClelland, J.W.; Holmes, R.M. Circumpolar synchrony in big river bacterioplankton. Proc. Natl. Acad. Sci. USA 2009, 106, 21208–21212. [Google Scholar] [CrossRef]
- Fan, L.M.; Song, C.; Meng, S.L.; Qiu, L.P.; Zheng, Y.; Wu, W.; Qu, J.H.; Li, D.D.; Zhang, C.; Hu, G.D.; et al. Spatial distribution of planktonic bacterial and archaeal communities in the upper section of the tidal reach in Yangtze River. Sci. Rep. 2016, 6, 39147. [Google Scholar]
- Sun, W.; Xia, C.Y.; Xu, M.Y.; Guo, J.; Sun, G.P. Seasonality Affects the Diversity and Composition of Bacterioplankton Communities in Dongjiang River, a Drinking Water Source of Hong Kong. Front. Microbiol. 2017, 8, 1644. [Google Scholar] [CrossRef]
- Adhikari, N.P.; Liu, Y.Q.; Liu, K.S.; Zhang, F.; Adhikari, S.; Chen, Y.Y.; Liu, X.B. Bacterial community composition and diversity in Koshi River, the largest river of Nepal. Ecol. Indic. 2019, 104, 501–511. [Google Scholar] [CrossRef]
- Dang, H.Y.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [PubMed]
- Allgaier, M.; Bruckner, S.; Jaspers, E.; Grossart, H.P. Intra- and inter-lake variability of free-living and particle-associated Actinobacteria communities. Environ. Microbiol. 2007, 9, 2728–2741. [Google Scholar] [PubMed]
- Elifantz, H.; Malmstrom, R.R.; Cottrell, M.T.; Kirchman, D.L. Assimilation of polysaccharides and glucose by major bacterial groups in the Delaware Estuary. Appl. Environ. Microbiol. 2005, 71, 7799–7805. [Google Scholar] [PubMed]
- Huang, Y.; Zou, L.; Zhang, S.Y.; Xie, S.G. Comparison of Bacterioplankton Communities in Three Heavily Polluted Streams in China. Biomed. Environ. Sci. 2011, 24, 140–145. [Google Scholar] [PubMed]
- Jeffries, T.C.; Fontes, M.L.S.; Harrison, D.P.; Van-Dongen-Vogels, V.; Eyre, B.D.; Ralph, P.J.; Seymour, J.R. Bacterioplankton Dynamics within a Large Anthropogenically Impacted Urban Estuary. Front. Microbiol. 2016, 6, 1438. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Z.; Gao, Y.C.; Huang, X.N.; Ni, P.; Wu, Y.N.; Deng, Y.; Zhan, A.B. Adaptive shifts of bacterioplankton communities in response to nitrogen enrichment in a highly polluted river. Environ. Pollut. 2019, 245, 290–299. [Google Scholar]
- Cotner, J.B.; Ammerman, J.W.; Peele, E.R.; Bentzen, E. Phosphorus-limited bacterioplankton growth in the Sargasso Sea. Aquat. Microb. Ecol. 1997, 13, 141–149. [Google Scholar] [CrossRef]
- Niu, Y.; Shen, H.; Chen, J.; Xie, P.; Yang, X.; Tao, M.; Ma, Z.; Qi, M. Phytoplankton community succession shaping bacterioplankton community composition in Lake Taihu, China. Water Res. 2011, 45, 4169–4182. [Google Scholar]
- Spietz, R.L.; Williams, C.M.; Rocap, G.; Horner-Devine, M.C. A Dissolved Oxygen Threshold for Shifts in Bacterial Community Structure in a Seasonally Hypoxic Estuary. PLoS ONE 2015, 10, e0135731. [Google Scholar]
- Ruiz-Gonzalez, C.; Proia, L.; Ferrera, I.; Gasol, J.M.; Sabater, S. Effects of large river dam regulation on bacterioplankton community structure. FEMS Microbiol. Ecol. 2013, 84, 316–331. [Google Scholar] [CrossRef]
- Walters, S.P.; Thebo, A.L.; Boehm, A.B. Impact of urbanization and agriculture on the occurrence of bacterial pathogens and stx genes in coastal waterbodies of central California. Water Res. 2011, 45, 1752–1762. [Google Scholar] [PubMed]
- Ibekwe, A.M.; Ma, J.; Murinda, S.E. Bacterial community composition and structure in an Urban River impacted by different pollutant sources. Sci. Total. Environ. 2016, 566, 1176–1185. [Google Scholar] [CrossRef]
- Zhen, G.; Li, Y.; Tong, Y.; Yang, L.; Zhu, Y.; Zhang, W. Temporal variation and regional transfer of heavy metals in the Pearl (Zhujiang) River, China. Environ. Sci. Pollut. Res. 2016, 23, 8410–8420. [Google Scholar]
- Fu, J.M.; Mai, B.X.; Sheng, G.Y.; Zhang, G.; Wang, X.M.; Peng, P.A.; Xiao, X.M.; Ran, R.; Cheng, F.Z.; Peng, X.Z.; et al. Persistent organic pollutants in environment of the Pearl River Delta, China: An overview. Chemosphere 2003, 52, 1411–1422. [Google Scholar] [CrossRef]
- Yu, J.A.; Ho, W.T.; Lu, H.M.; Yang, Y.F. Study on water quality and genotoxicity of surface microlayer and subsurface water in Guangzhou section of Pearl River. Environ. Monit. Assess. 2011, 174, 681–692. [Google Scholar]
- Strokal, M.; Kroeze, C.; Li, L.L.; Luan, S.J.; Wang, H.Z.; Yang, S.S.; Zhang, Y.S. Increasing dissolved nitrogen and phosphorus export by the Pearl River (Zhujiang): A modeling approach at the sub-basin scale to assess effective nutrient management. Biogeochemistry 2015, 125, 221–242. [Google Scholar] [CrossRef]
- Jiao, S.; Liu, Z.S.; Lin, Y.B.; Yang, J.; Chen, W.M.; Wei, G.H. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Lopes, F.A.; Catao, E.C.; Santana, R.H.; Cabral Ade, S.; Paranhos, R.; Rangel, T.P.; de Rezende, C.E.; Edwards, R.A.; Thompson, C.C.; Thompson, F.L.; et al. Microbial Community Profile and Water Quality in a Protected Area of the Caatinga Biome. PLoS ONE 2016, 11, e0148296. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tests | Group | R Value | p Value |
|---|---|---|---|
| Global test | BJ–XJ–PRD | 0.835 | 0.001 |
| Paired comparison | BJ–XJ | 0.998 | 0.001 |
| BJ–PRD | 0.979 | 0.001 | |
| XJ–PRD | 0.385 | 0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Chen, W.; Sun, J.; Liu, L.; Huang, X. Spatial Variation in Bacterioplankton Communities in the Pearl River, South China: Impacts of Land Use and Physicochemical Factors. Microorganisms 2020, 8, 814. https://doi.org/10.3390/microorganisms8060814
Zhou L, Chen W, Sun J, Liu L, Huang X. Spatial Variation in Bacterioplankton Communities in the Pearl River, South China: Impacts of Land Use and Physicochemical Factors. Microorganisms. 2020; 8(6):814. https://doi.org/10.3390/microorganisms8060814
Chicago/Turabian StyleZhou, Lei, Weiyuan Chen, Jijia Sun, Li Liu, and Xiande Huang. 2020. "Spatial Variation in Bacterioplankton Communities in the Pearl River, South China: Impacts of Land Use and Physicochemical Factors" Microorganisms 8, no. 6: 814. https://doi.org/10.3390/microorganisms8060814
APA StyleZhou, L., Chen, W., Sun, J., Liu, L., & Huang, X. (2020). Spatial Variation in Bacterioplankton Communities in the Pearl River, South China: Impacts of Land Use and Physicochemical Factors. Microorganisms, 8(6), 814. https://doi.org/10.3390/microorganisms8060814