Comparing the Influence of Assembly Processes Governing Bacterial Community Succession Based on DNA and RNA Data



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Site and Soil Sampling
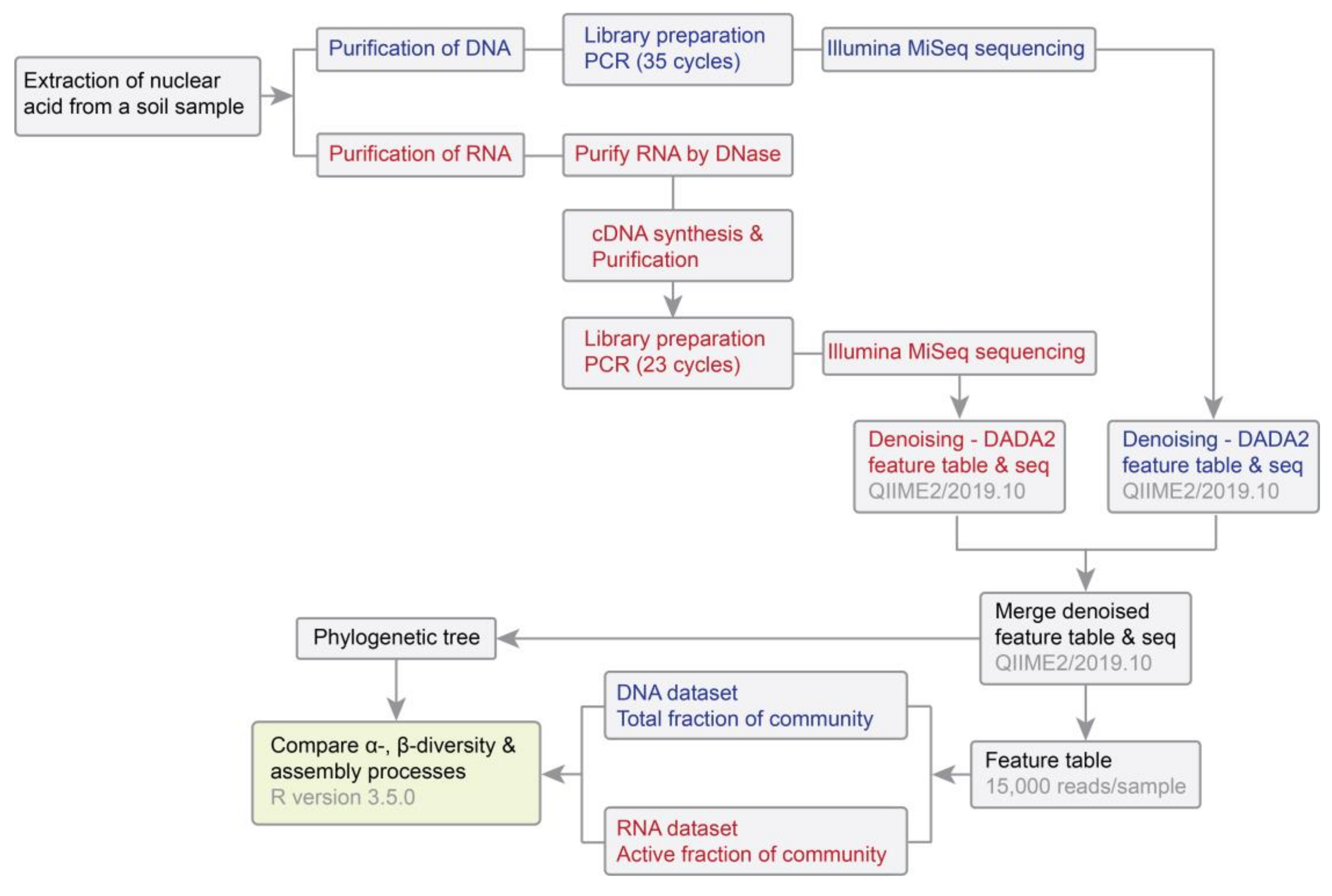
2.2. Nucleic Acid Extraction, Amplicon Library Preparation and Sequencing
2.3. Sequence Processing, Analysis of Community Structure and Statistical Analyses
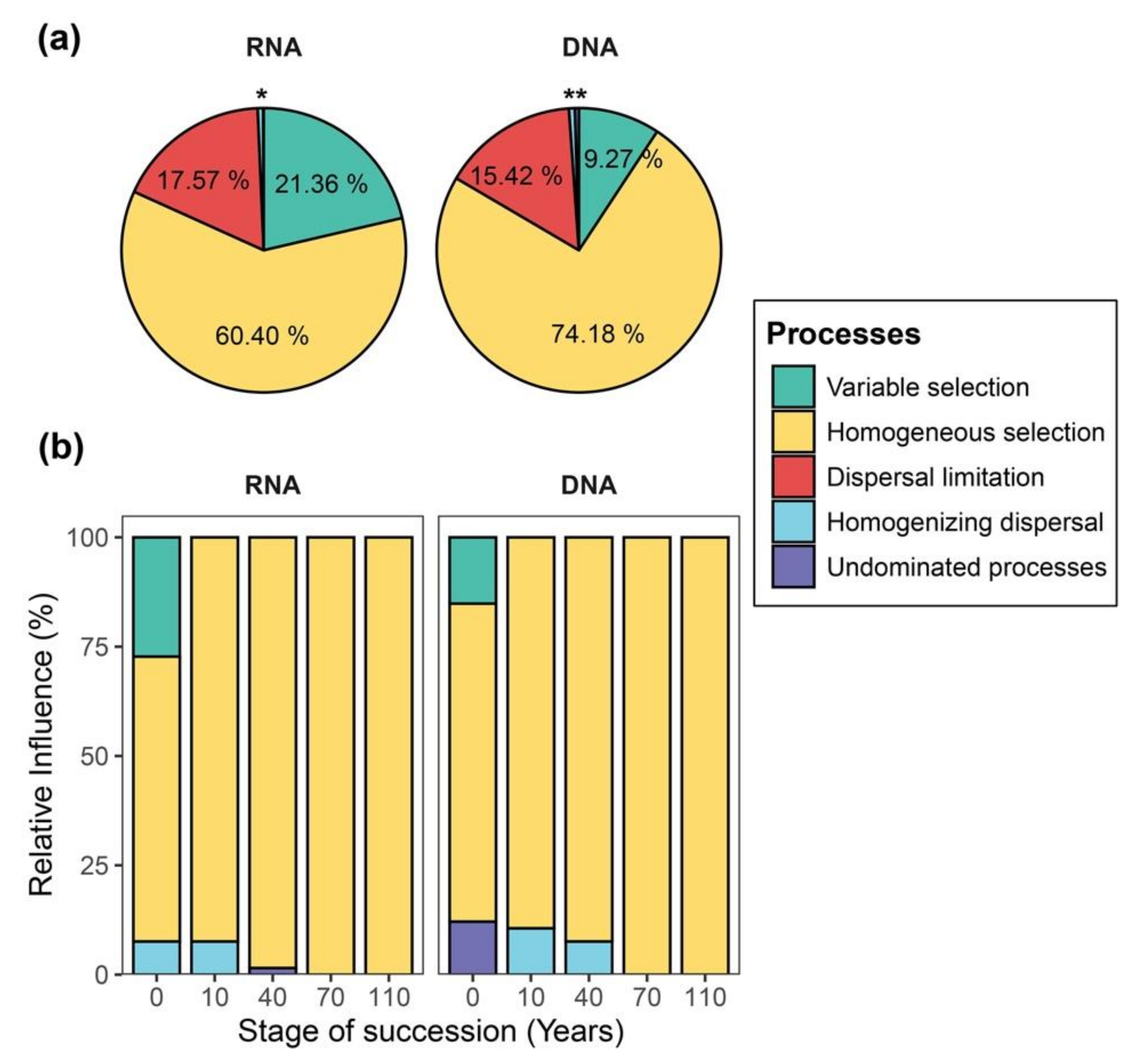
2.4. Quantification of Community Assembly Processes Governing Community Succession
3. Results
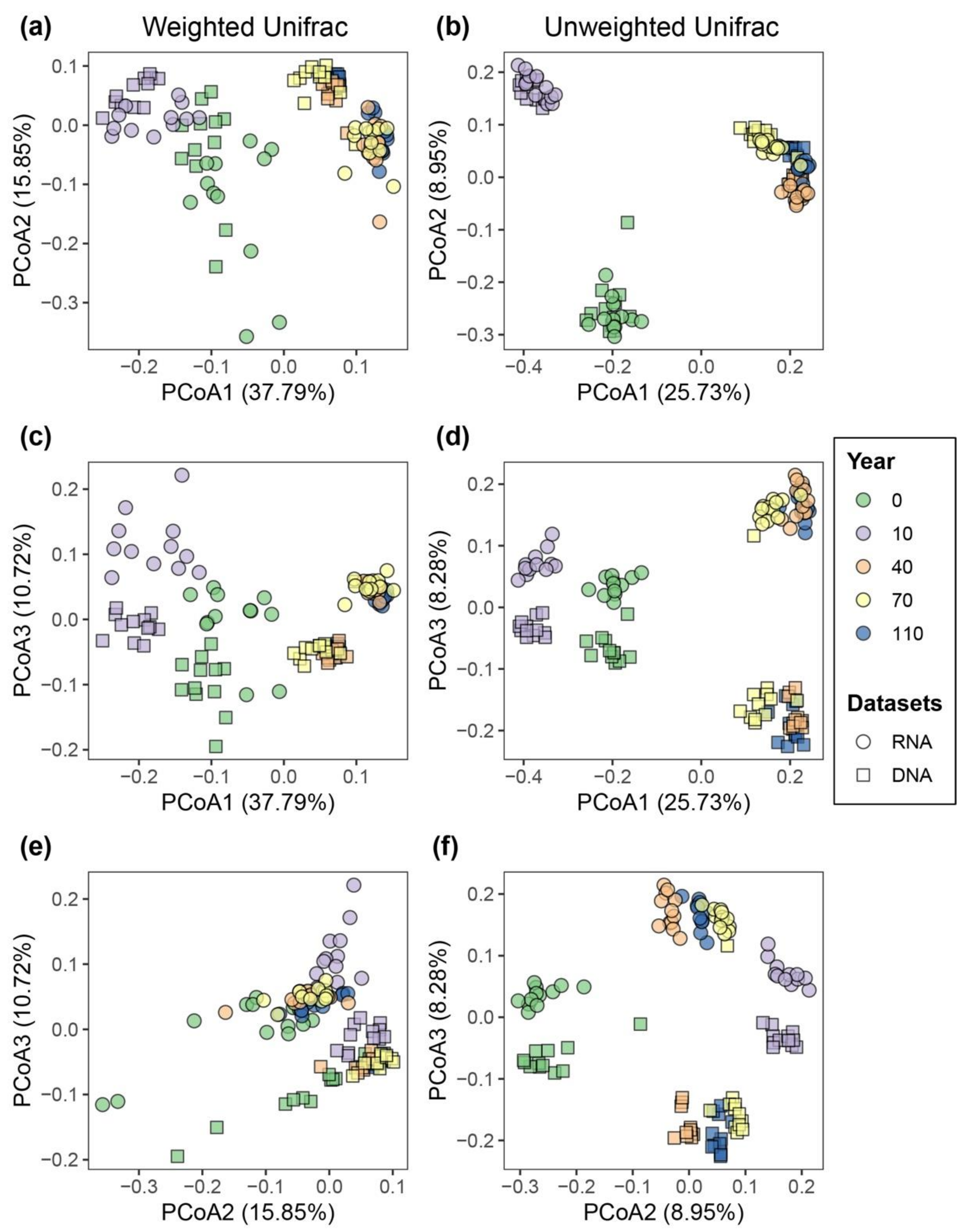
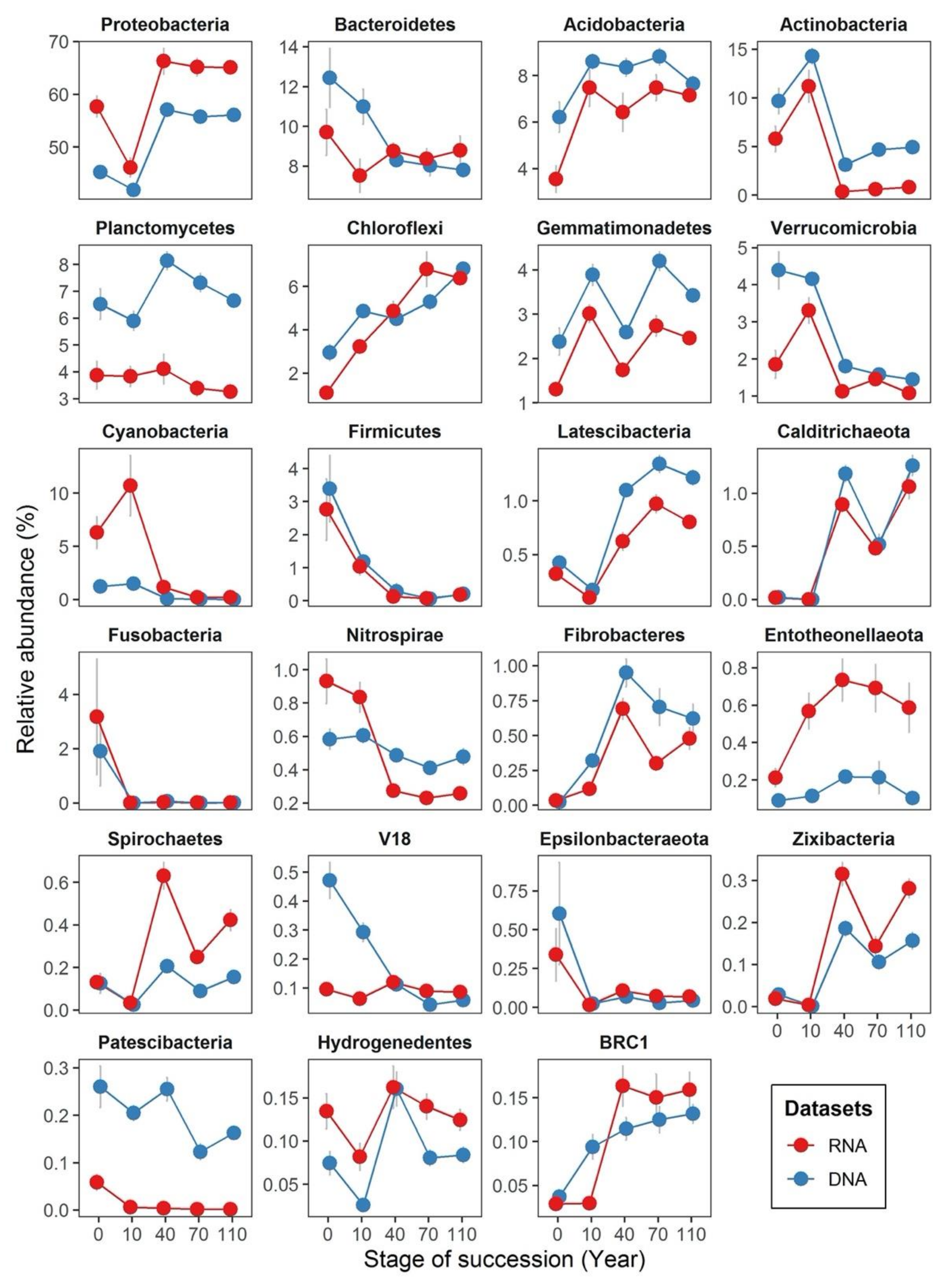
3.1. Comparing Bacterial Community Structure Based on DNA and RNA Approaches
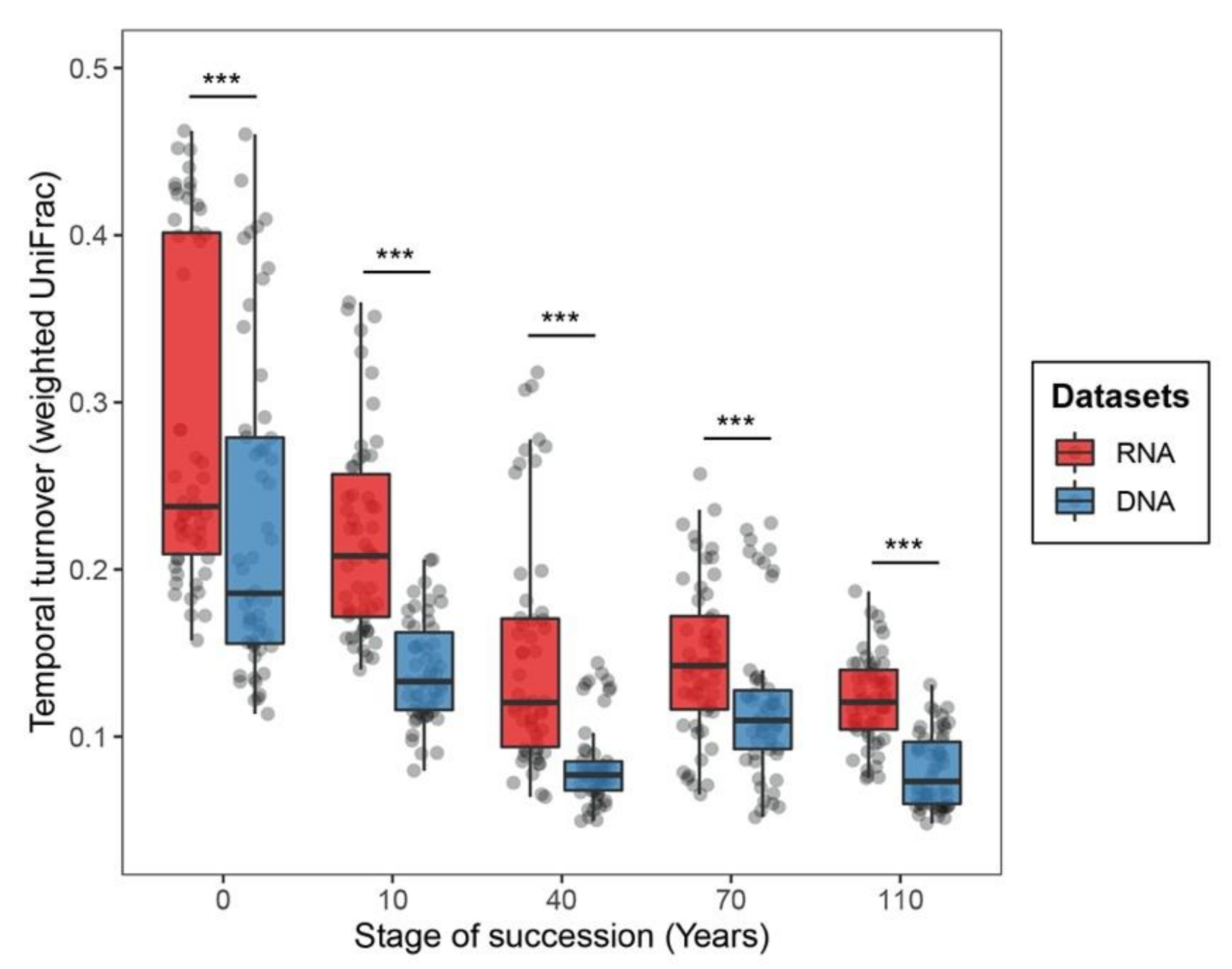
3.2. Differences in Aseembly Processes between DNA- and RNA-Inferred Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burcham, Z.M.; Pechal, J.L.; Schmidt, C.J.; Bose, J.L.; Rosch, J.W.; Benbow, M.E.; Jordan, H.R. Bacterial Community Succession, Transmigration, and Differential Gene Transcription in a Controlled Vertebrate Decomposition Model. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Anderson, S.P.; Cleveland, C.C.; Martin, A.P.; Miller, A.E.; Seimon, A.; Schmidt, S.K. Microbial community succession in an unvegetated, recently deglaciated soil. Microb. Ecol. 2007, 53, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Jumpponen, A. Contrasting primary successional trajectories of fungi and bacteria in retreating glacier soils. Mol. Ecol. 2014, 23, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Castle, S.C.; Nemergut, D.R.; Grandy, A.S.; Leff, J.W.; Graham, E.B.; Hood, E.; Schmidt, S.K.; Wickings, K.; Cleveland, C.C. Biogeochemical drivers of microbial community convergence across actively retreating glaciers. Soil Biol. Biochem. 2016, 101, 74–84. [Google Scholar] [CrossRef]
- Vigneron, A.; Alsop, E.B.; Lomans, B.P.; Kyrpides, N.C.; Head, I.M.; Tsesmetzis, N. Succession in the petroleum reservoir microbiome through an oil field production lifecycle. ISME J. 2017, 11, 2141–2154. [Google Scholar] [CrossRef]
- Lorenz, M.G.; Wackernagel, W. Adsorption of DNA to sand and variable degradation rates of adsorbed DNA. Appl. Environ. Microbiol. 1987, 53, 2948. [Google Scholar] [CrossRef]
- Carini, P.; Marsden, P.J.; Leff, J.W.; Morgan, E.E.; Strickland, M.S.; Fierer, N. Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2016, 2, 16242. [Google Scholar] [CrossRef]
- Emerson, J.B.; Adams, R.I.; Roman, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Schrodinger’s microbes: Tools for Distinguishing the living from the dead in microbial ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef]
- Carini, P.; Delgado-Baquerizo, M.; Hinckley, E.-L.S.; Holland-Moritz, H.; Brewer, T.E.; Rue, G.; Vanderburgh, C.; McKnight, D.; Fierer, N. Effects of Spatial Variability and Relic DNA Removal on the Detection of Temporal Dynamics in Soil Microbial Communities. mBio 2020, 11, e02776-19. [Google Scholar] [CrossRef]
- Lennon, J.T.; Muscarella, M.E.; Placella, S.A.; Lehmkuhl, B.K. How, When, and Where Relic DNA Affects Microbial Diversity. mBio 2018, 9, e00637-18. [Google Scholar] [CrossRef] [PubMed]
- Schippers, A.; Neretin, L.N.; Kallmeyer, J.; Ferdelman, T.G.; Cragg, B.A.; John Parkes, R.; Jørgensen, B.B. Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 2005, 433, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Denef, V.J.; Fujimoto, M.; Berry, M.A.; Schmidt, M.L. Seasonal Succession Leads to Habitat-Dependent Differentiation in Ribosomal RNA:DNA Ratios among Freshwater Lake Bacteria. Front. Microbiol. 2016, 7, 606. [Google Scholar] [CrossRef] [PubMed]
- Jurburg, S.D.; Nunes, I.; Brejnrod, A.; Jacquiod, S.; Prieme, A.; Sorensen, S.J.; Van Elsas, J.D.; Salles, J.F. Legacy Effects on the Recovery of Soil Bacterial Communities from Extreme Temperature Perturbation. Front. Microbiol. 2017, 8, 1832. [Google Scholar] [CrossRef]
- Jurburg, S.D.; Nunes, I.; Stegen, J.C.; Le Roux, X.; Prieme, A.; Sorensen, S.J.; Salles, J.F. Autogenic succession and deterministic recovery following disturbance in soil bacterial communities. Sci. Rep. 2017, 7, 45691. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef]
- Ferrenberg, S.; O’Neill, S.P.; Knelman, J.E.; Todd, B.; Duggan, S.; Bradley, D.; Robinson, T.; Schmidt, S.K.; Townsend, A.R.; Williams, M.W.; et al. Changes in assembly processes in soil bacterial communities following a wildfire disturbance. ISME J. 2013, 7, 1102–1111. [Google Scholar] [CrossRef]
- Mueller, R.C.; Gallegos-Graves, L.; Zak, D.R.; Kuske, C.R. Assembly of Active Bacterial and Fungal Communities Along a Natural Environmental Gradient. Microb Ecol 2016, 71, 57–67. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Schrama, M.; Berg Mp Fau - Olff, H.; Olff, H. Ecosystem assembly rules: the interplay of green and brown webs during salt marsh succession. Ecology 2012, 93(11), 2353–2364. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Silva, M.; Triado-Margarit, X.; Casamayor, E.O.; van Elsas, J.D.; Salles, J.F. Dynamics of bacterial community succession in a salt marsh chronosequence: evidences for temporal niche partitioning. ISME J. 2014, 8, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–648. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed]
- Chase, J.M.; Kraft, N.J.B.; Smith, K.G.; Vellend, M.; Inouye, B.D. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2011, 2, art24. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: elegant graphics for data analysis. J. Stat. Softw. 2010, 35, 65–88. [Google Scholar]
- Ortiz-Alvarez, R.; Fierer, N.; de Los Rios, A.; Casamayor, E.O.; Barberan, A. Consistent changes in the taxonomic structure and functional attributes of bacterial communities during primary succession. ISME J. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.I.; Whiteley, A.S.; Donnell, A.G.; Bailey, M.J. Rapid Method for Coextraction of DNA and RNA from Natural Environments for Analysis of Ribosomal DNA- and rRNA-Based Microbial Community Composition. Appl. Environ. Microbiol. 2000, 66, 5488. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.M.; Petersen, I.A.B.; Tobi, E.; Korte, L.; Bohannan, B.J.M. Use of RNA and DNA to Identify Mechanisms of Bacterial Community Homogenization. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Bastida, F.; Torres, I.F.; Andres-Abellan, M.; Baldrian, P.; Lopez-Mondejar, R.; Vetrovsky, T.; Richnow, H.H.; Starke, R.; Ondono, S.; Garcia, C.; et al. Differential sensitivity of total and active soil microbial communities to drought and forest management. Glob. Chang. Biol. 2017, 23, 4185–4203. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Lankiewicz, T.S.; Cottrell, M.T.; Kirchman, D.L. Growth rates and rRNA content of four marine bacteria in pure cultures and in the Delaware estuary. ISME J. 2016, 10, 823–832. [Google Scholar] [CrossRef]
- Sukenik, A.; Kaplan-Levy, R.N.; Welch, J.M.; Post, A.F. Massive multiplication of genome and ribosomes in dormant cells (akinetes) of Aphanizomenon ovalisporum (Cyanobacteria). ISME J. 2012, 6, 670–679. [Google Scholar] [CrossRef]
- Nawaz, A.; Purahong, W.; Herrmann, M.; Kusel, K.; Buscot, F.; Wubet, T. DNA- and RNA- Derived Fungal Communities in Subsurface Aquifers Only Partly Overlap but React Similarly to Environmental Factors. Microorganisms 2019, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.; Mølbak, L.; Molin, S. Bacterial Activity in the Rhizosphere Analyzed at the Single-Cell Level by Monitoring Ribosome Contents and Synthesis Rates. Appl. Environ. Microbiol. 2000, 66, 801. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, L.; Besemer, K.; Fasching, C.; Urich, T.; Singer, G.A.; Quince, C.; Battin, T.J. Rare but active taxa contribute to community dynamics of benthic biofilms in glacier-fed streams. Environ. Microbiol. 2014, 16, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Lennon, J.T. Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. USA 2010, 107, 5881–5886. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Df * | SumSqs * | MeanSqs * | F.Model * | R2 * | p-Values † |
|---|---|---|---|---|---|---|
| Dataset | 1 | 0.605792 | 0.605792 | 65.36243 | 0.123029 | <0.001 |
| Year | 4 | 2.373268 | 0.593317 | 64.01644 | 0.481983 | <0.001 |
| Month | 3 | 0.164918 | 0.054973 | 5.931321 | 0.033493 | <0.001 |
| Dataset:Year | 4 | 0.309029 | 0.077257 | 8.335727 | 0.06276 | <0.001 |
| Dataset:Month | 3 | 0.047992 | 0.015997 | 1.72604 | 0.009747 | 0.0436 |
| Year:Month | 12 | 0.523401 | 0.043617 | 4.706062 | 0.106297 | <0.001 |
| Dataset:Year:Month | 12 | 0.158108 | 0.013176 | 1.421602 | 0.03211 | 0.0246 |
| Residuals | 80 | 0.741456 | 0.009268 | 0.150581 | ||
| Total | 119 | 4.923963 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; Dini-Andreote, F.; Falcão Salles, J. Comparing the Influence of Assembly Processes Governing Bacterial Community Succession Based on DNA and RNA Data. Microorganisms 2020, 8, 798. https://doi.org/10.3390/microorganisms8060798
Jia X, Dini-Andreote F, Falcão Salles J. Comparing the Influence of Assembly Processes Governing Bacterial Community Succession Based on DNA and RNA Data. Microorganisms. 2020; 8(6):798. https://doi.org/10.3390/microorganisms8060798
Chicago/Turabian StyleJia, Xiu, Francisco Dini-Andreote, and Joana Falcão Salles. 2020. "Comparing the Influence of Assembly Processes Governing Bacterial Community Succession Based on DNA and RNA Data" Microorganisms 8, no. 6: 798. https://doi.org/10.3390/microorganisms8060798
APA StyleJia, X., Dini-Andreote, F., & Falcão Salles, J. (2020). Comparing the Influence of Assembly Processes Governing Bacterial Community Succession Based on DNA and RNA Data. Microorganisms, 8(6), 798. https://doi.org/10.3390/microorganisms8060798