Comparative Genomics of the Rhodococcus Genus Shows Wide Distribution of Biodegradation Traits



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Phylogenomic Analysis
2.3. Clustering of Rhodococcus Genomes
2.4. Orthologous Groups Identification and Genome Fractions
2.5. Phylogeny of Single-Copy Genes
2.6. Diversity of Rieske 2Fe-2S Dioxygenases
3. Results and Discussion
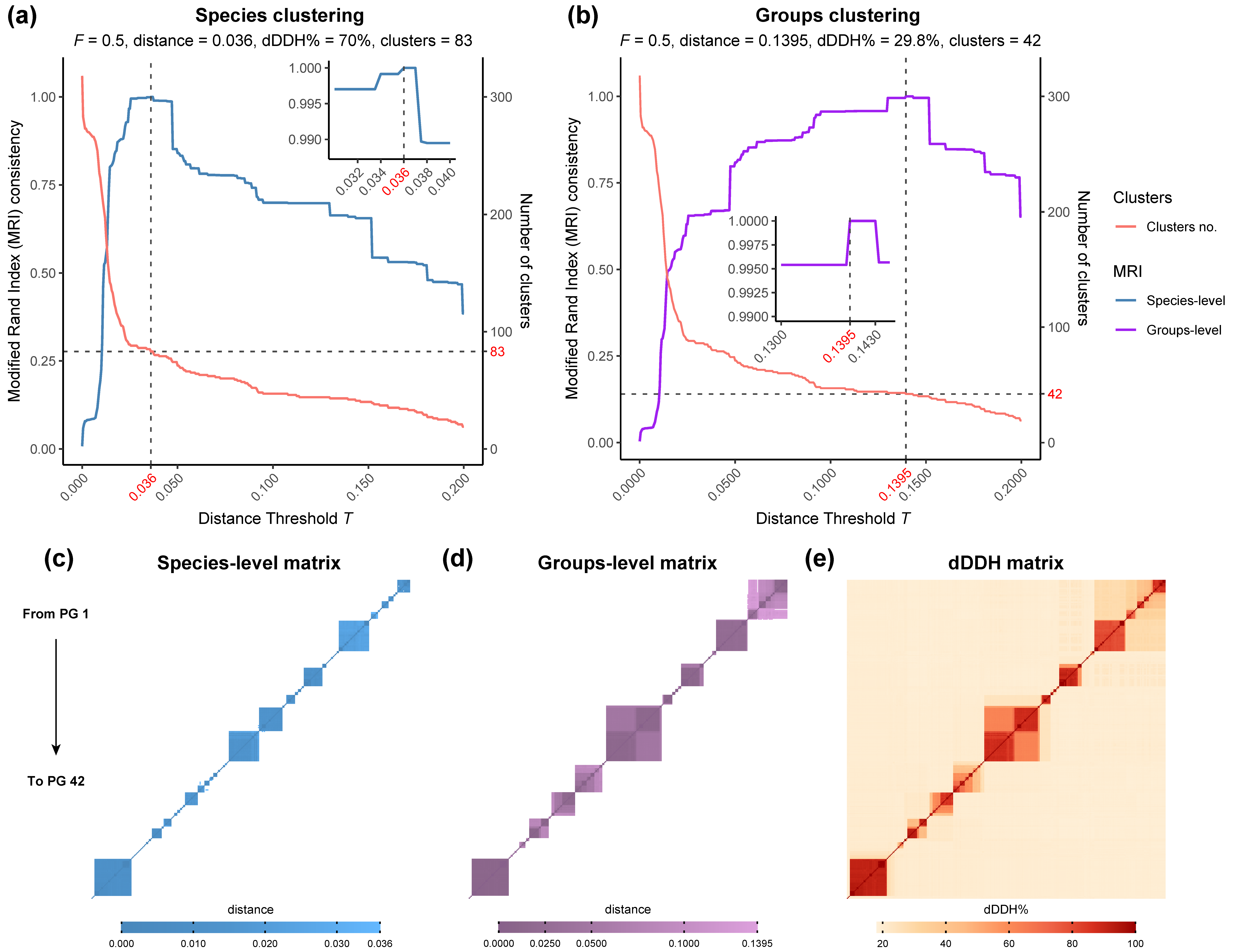
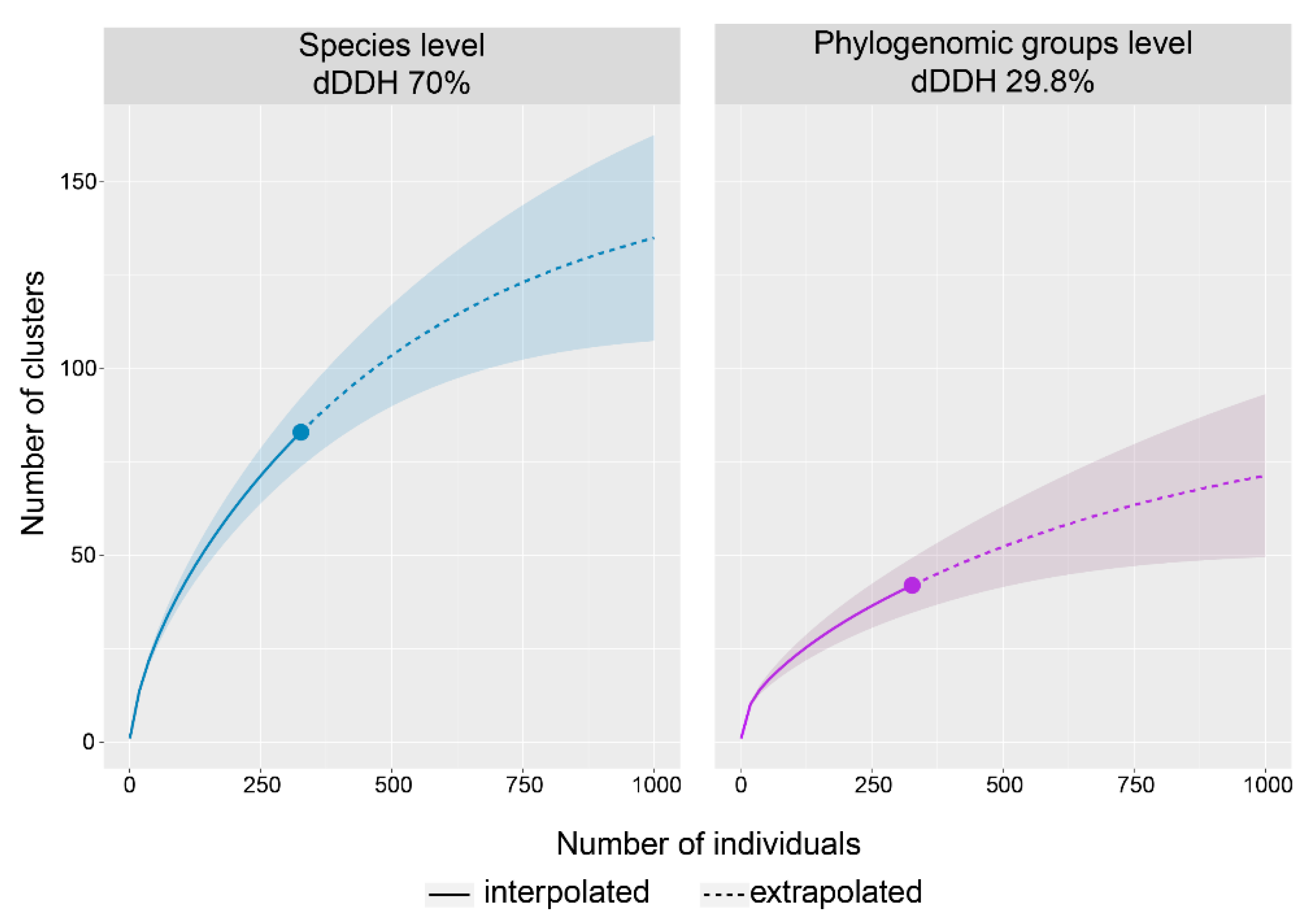
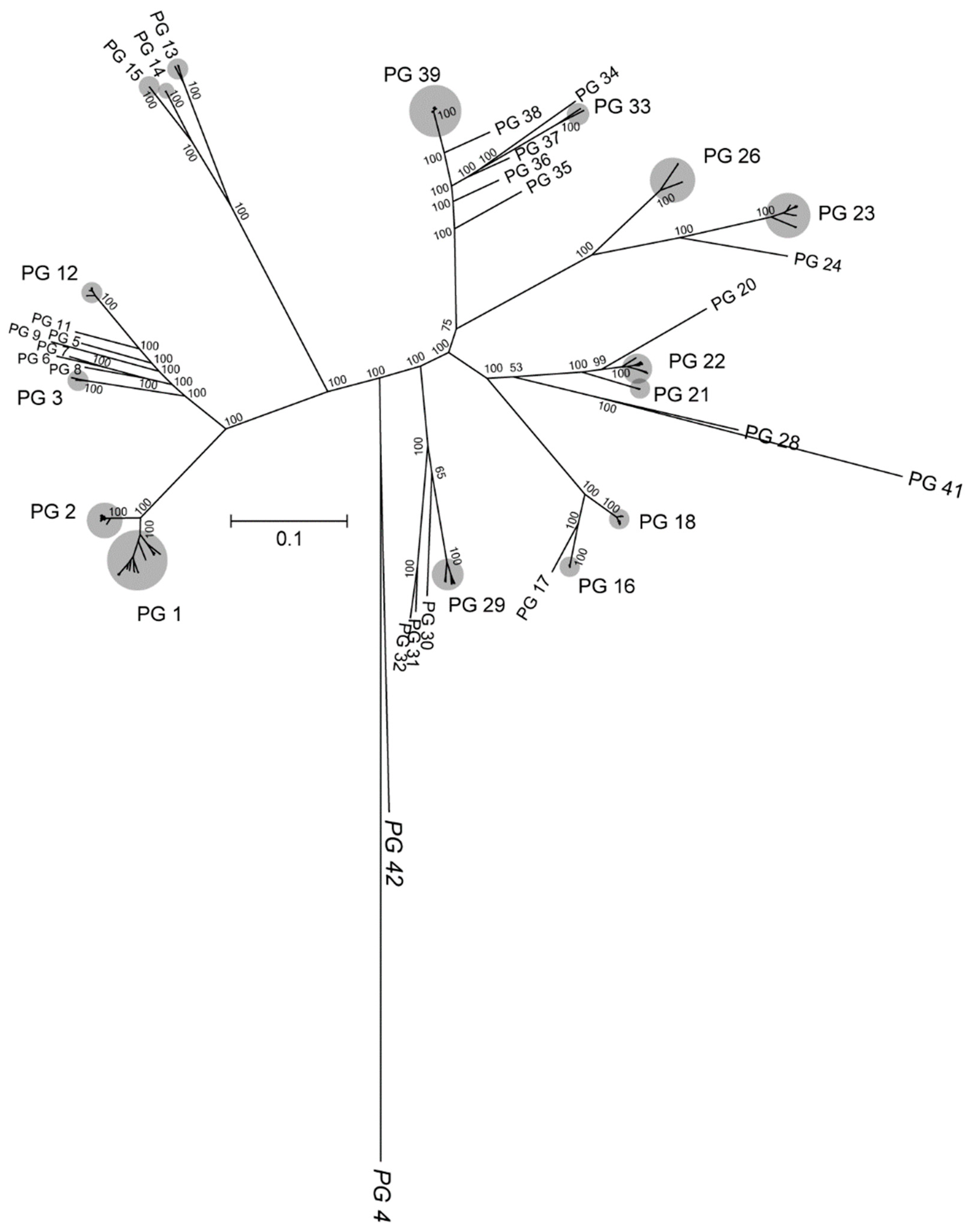
3.1. Phylogenomic Analysis and Clustering of the Rhodococcus Genus
3.2. Phylogeny Based on Single-Copy Proteins
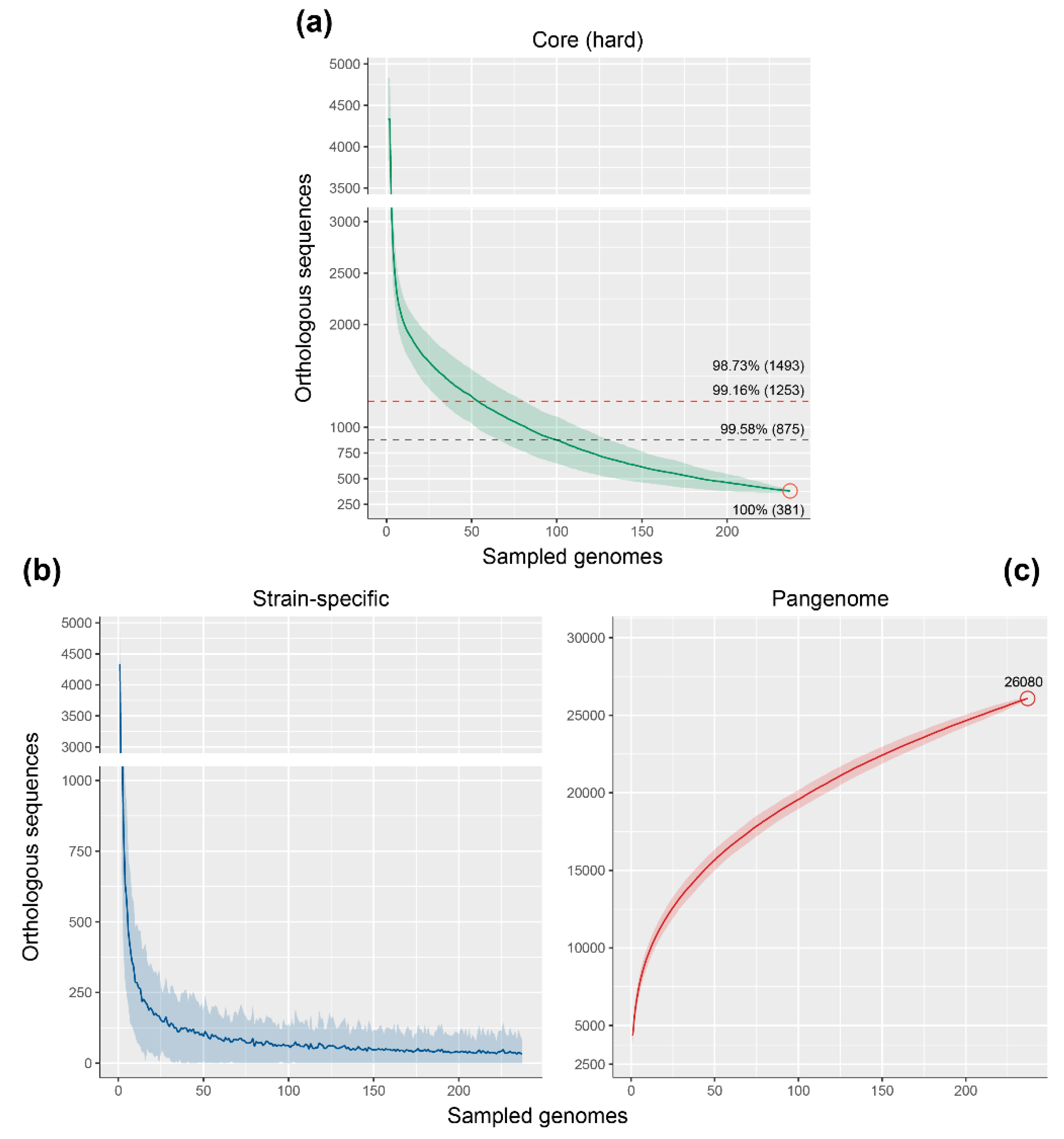
3.3. Genome Fractions of the Rhodococcus Genus
3.4. Distribution of PAHs and Alkane Degradation Genes
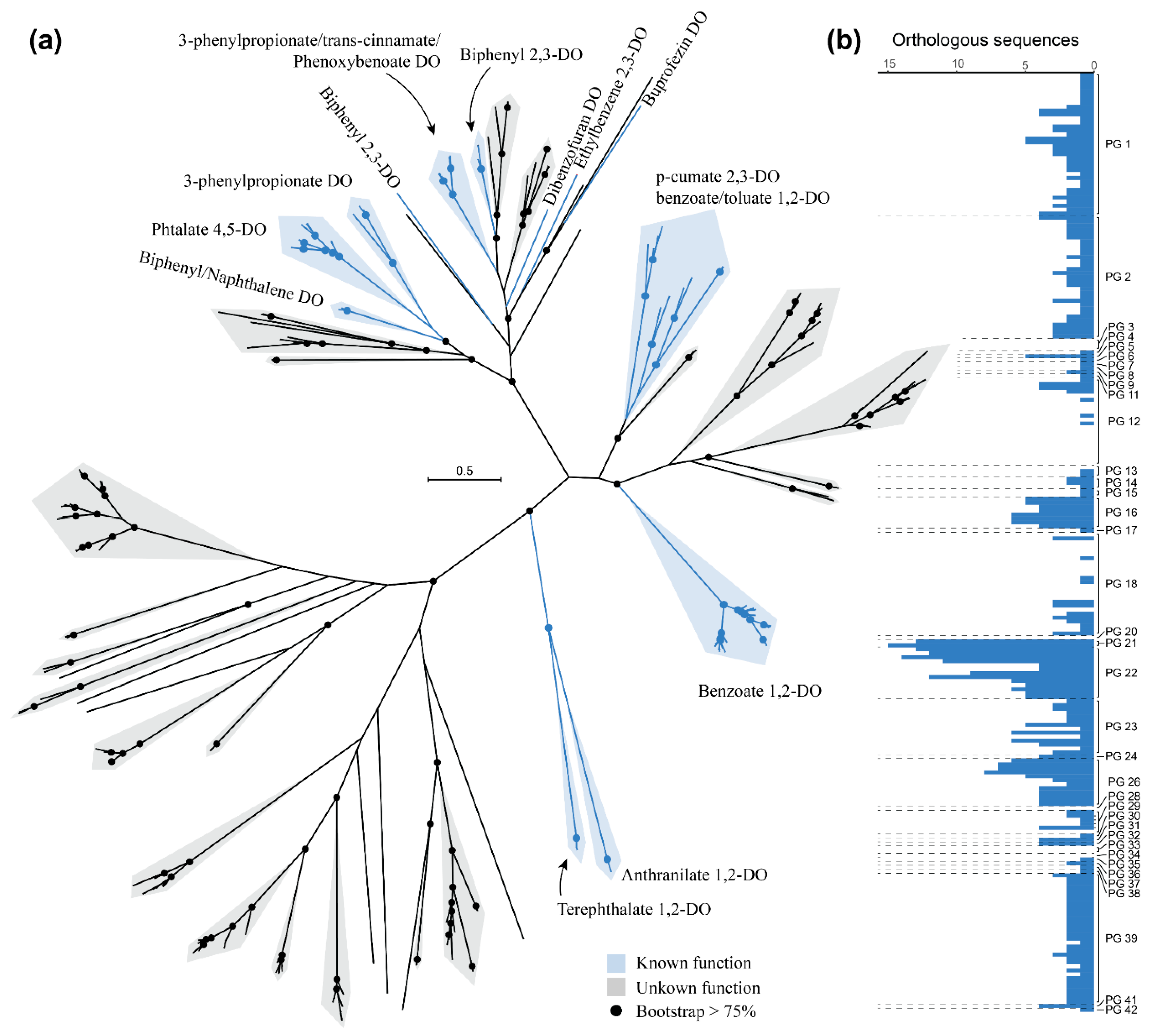
3.5. Diversity of Rieske 2Fe-2S Dioxygenases among Rhodococcus Genomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Helmke, E.; Weyland, H. Rhodococcus marinonascens sp. nov., an actinomycete from the sea. Int. J. Syst. Evol. Microbiol. 1984, 34, 127–138. [Google Scholar] [CrossRef]
- Margesin, R.; Labbe, D.; Schinner, F.; Greer, C.; Whyte, L. Characterization of hydrocarbon-degrading microbial populations in contaminated and pristine alpine soils. Appl. Environ. Microbiol. 2003, 69, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-W.; Joo, Y.-H.; An, Y.-J.; Cho, K.-S. Isolation and characterization of psychrotrophic and halotolerant Rhodococcus sp. YHLT-2. J. Microbiol. Biotechnol. 2006, 16, 605–612. [Google Scholar]
- Adnani, N.; Braun, D.R.; McDonald, B.R.; Chevrette, M.G.; Currie, C.R.; Bugni, T.S. Complete genome sequence of Rhodococcus sp. strain WMMA185, a marine sponge-associated bacterium. Genome Announc. 2016, 4, e01406–e01416. [Google Scholar] [CrossRef]
- Yassin, A. Rhodococcus triatomae sp. nov., isolated from a blood-sucking bug. Int. J. Syst. Evol. Microbiol. 2005, 55, 1575–1579. [Google Scholar] [CrossRef]
- Giguère, S.; Cohen, N.; Keith Chaffin, M.; Hines, S.; Hondalus, M.; Prescott, J.; Slovis, N. Rhodococcus equi: Clinical Manifestations, Virulence, and Immunity. J. Vet. Intern. Med. 2011, 25, 1221–1230. [Google Scholar] [CrossRef]
- Prescott, J.F. Rhodococcus equi: An animal and human pathogen. Clin. Microbiol. Rev. 1991, 4, 20–34. [Google Scholar] [CrossRef]
- Cornelis, K.; Ritsema, T.; Nijsse, J.; Holsters, M.; Goethals, K.; Jaziri, M. The plant pathogen Rhodococcus fascians colonizes the exterior and interior of the aerial parts of plants. Mol. Plant-Microbe Interact. 2001, 14, 599–608. [Google Scholar] [CrossRef]
- Goethals, K.; Vereecke, D.; Jaziri, M.; Van Montagu, M.; Holsters, M. Leafy gall formation by Rhodococcus fascians. Annu. Rev. Phytopathol. 2001, 39, 27–52. [Google Scholar] [CrossRef]
- De Carvalho, C.C.; Parreño-Marchante, B.; Neumann, G.; Da Fonseca, M.M.R.; Heipieper, H.J. Adaptation of Rhodococcus erythropolis DCL14 to growth on n-alkanes, alcohols and terpenes. Appl. Microbiol. Biotechnol. 2005, 67, 383–388. [Google Scholar] [CrossRef]
- Iwasaki, T.; Takeda, H.; Miyauchi, K.; Yamada, T.; Masai, E.; Fukuda, M. Characterization of two biphenyl dioxygenases for biphenyl/PCB degradation in a PCB degrader, Rhodococcus sp. strain RHA1. Biosci. Biotechnol. Biochem. 2007, 71, 993–1002. [Google Scholar] [CrossRef]
- Song, X.; Xu, Y.; Li, G.; Zhang, Y.; Huang, T.; Hu, Z. Isolation, characterization of Rhodococcus sp. P14 capable of degrading high-molecular-weight polycyclic aromatic hydrocarbons and aliphatic hydrocarbons. Mar. Pollut. Bull. 2011, 62, 2122–2128. [Google Scholar] [CrossRef] [PubMed]
- McLeod, M.P.; Warren, R.L.; Hsiao, W.W.; Araki, N.; Myhre, M.; Fernandes, C.; Miyazawa, D.; Wong, W.; Lillquist, A.L.; Wang, D. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. USA 2006, 103, 15582–15587. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Kim, Y.-J.; Kim, I.S. Rhodococcus subtropicus sp. nov., a new actinobacterium isolated from a cave. Int. J. Syst. Evol. Microbiol. 2019, 69, 3128–3134. [Google Scholar] [CrossRef]
- Silva, L.J.; Souza, D.T.; Genuario, D.B.; Hoyos, H.A.V.; Santos, S.N.; Rosa, L.H.; Zucchi, T.D.; Melo, I.S. Rhodococcus psychrotolerans sp. nov., isolated from rhizosphere of Deschampsia antarctica. Antonie Van Leeuwenhoek 2018, 111, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L.; Zhang, X.; Zhang, S.; Yang, L.; Yuan, H.; Chen, J.; Liang, C.; Huang, W.; Liu, J. Rhodococcus daqingensis sp. nov., isolated from petroleum-contaminated soil. Antonie van Leeuwenhoek 2019, 112, 695–702. [Google Scholar] [CrossRef]
- Kämpfer, P.; Dott, W.; Martin, K.; Glaeser, S.P. Rhodococcus defluvii sp. nov., isolated from wastewater of a bioreactor and formal proposal to reclassify [Corynebacterium hoagii] and Rhodococcus equi as Rhodococcus hoagii comb. nov. Int. J. Syst. Evol. Microbiol. 2014, 64, 755–761. [Google Scholar] [CrossRef]
- Tindall, B. A note on the genus name Rhodococcus Zopf 1891 and its homonyms. Int. J. Syst. Evol. Microbiol. 2014, 64, 1062–1064. [Google Scholar] [CrossRef]
- Jones, A.; Sutcliffe, I.; Goodfellow, M. Proposal to replace the illegitimate genus name Prescottia Jones et al. 2013 with the genus name Prescottella gen. nov. and to replace the illegitimate combination Prescottia equi Jones et al. 2013 with Prescottella equi comb. nov. Antonie van Leeuwenhoek 2013, 103, 1405–1407. [Google Scholar] [CrossRef]
- Sangal, V.; Goodfellow, M.; Jones, A.L.; Seviour, R.J.; Sutcliffe, I.C. Refined Systematics of the Genus Rhodococcus Based on Whole Genome Analyses. In Biology of Rhodococcus; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–21. [Google Scholar]
- Tindall, B. The correct name of the taxon that contains the type strain of Rhodococcus equi. Int. J. Syst. Evol. Microbiol. 2014, 64, 302–308. [Google Scholar] [CrossRef]
- Parte, A.C. LPSN—list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed]
- Orro, A.; Cappelletti, M.; D’Ursi, P.; Milanesi, L.; Di Canito, A.; Zampolli, J.; Collina, E.; Decorosi, F.; Viti, C.; Fedi, S. Genome and phenotype microarray analyses of Rhodococcus sp. BCP1 and Rhodococcus opacus R7: Genetic determinants and metabolic abilities with environmental relevance. PLOS ONE 2015, 10, e0139467. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, E.; MacArthur, I.; Scortti, M.; Alvarez, S.; Giguère, S.; Vázquez-Boland, J.A. Pangenome and phylogenomic analysis of the pathogenic actinobacterium Rhodococcus equi. Genome Biol. Evol. 2016, 8, 3140–3148. [Google Scholar] [CrossRef] [PubMed]
- Creason, A.L.; Davis, E.W.; Putnam, M.L.; Vandeputte, O.M.; Chang, J.H. Use of whole genome sequences to develop a molecular phylogenetic framework for Rhodococcus fascians and the Rhodococcus genus. Front. Plant Sci. 2014, 5, 406. [Google Scholar] [CrossRef]
- Gürtler, V.; Mayall, B.C.; Seviour, R. Can whole genome analysis refine the taxonomy of the genus Rhodococcus? FEMS Microbiol. Rev. 2004, 28, 377–403. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Garrido-Sanz, D.; Meier-Kolthoff, J.P.; Göker, M.; Martin, M.; Rivilla, R.; Redondo-Nieto, M. Genomic and genetic diversity within the Pseudomonas fluorescens complex. PLOS ONE 2016, 11, e0150183. [Google Scholar] [CrossRef]
- Garrido-Sanz, D.; Redondo-Nieto, M.; Mongiardini, E.; Blanco-Romero, E.; Durán, D.; Quelas, J.I.; Martin, M.; Rivilla, R.; Lodeiro, A.R.; Althabegoiti, M.J. Phylogenomic analyses of Bradyrhizobium reveal uneven distribution of the lateral and subpolar flagellar systems, which extends to Rhizobiales. Microorganisms 2019, 7, 50. [Google Scholar] [CrossRef]
- Garrido-Sanz, D.; Manzano, J.; Martín, M.; Redondo-Nieto, M.; Rivilla, R. Metagenomic analysis of a biphenyl-degrading soil bacterial consortium reveals the metabolic roles of specific populations. Front. Microbiol. 2018, 9, 232. [Google Scholar] [CrossRef]
- Garrido-Sanz, D.; Sansegundo-Lobato, P.; Redondo-Nieto, M.; Suman, J.; Cajthaml, T.; Blanco-Romero, E.; Martin, M.; Uhlik, O.; Rivilla, R. Analysis of the biodegradative and adaptive potential of the novel polychlorinated biphenyl degrader Rhodococcus sp. WAY2 revealed by its complete genome sequence. Microb. Genom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Kitagawa, W.; Mori, T.; Nakashima, N.; Tamura, T.; Kamagata, Y. Genetic and biochemical characterization of the dioxygenase involved in lateral dioxygenation of dibenzofuran from Rhodococcus opacus strain SAO101. Appl. Microbiol. Biotechnol. 2006, 73, 474–484. [Google Scholar] [CrossRef] [PubMed]
- NCBI ftp Server. Available online: ftp://ftp.ncbi.nlm.nih.gov (accessed on 1 June 2019).
- Genome-to-genome Distance Calculator (GGDC) 2.1. Available online: http://ggdc.dsmz.de/ggdc.php (accessed on 1 July 2019).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Göker, M.; García-Blázquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A. Complete genome sequence of DSM 30083 T, the type strain (U5/41 T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdisciplinary Reviews. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1. Available online: https://cran.r-project.org/web/packages/pheatmap/ (accessed on 1 November 2019).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.; Moret, B.M.; Stamatakis, A. How many bootstrap replicates are necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Li, J.; Zhou, J.; Wu, Y.; Yang, S.; Tian, D. GC-content of synonymous codons profoundly influences amino acid usage. G3: Genes, Genomes, Genet. 2015, 5, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Zakham, F.; Aouane, O.; Ussery, D.; Benjouad, A.; Ennaji, M.M. Computational genomics-proteomics and Phylogeny analysis of twenty one mycobacterial genomes (Tuberculosis & non Tuberculosis strains). Microbial Inform. Exp. 2012, 2, 7. [Google Scholar]
- Kim, J.-N.; Kim, Y.; Jeong, Y.; Roe, J.-H.; Kim, B.-G.; Cho, B.-K. Comparative genomics reveals the core and accessory genomes of Streptomyces species. J. Microbiol. Biotechnol. 2015, 25, 1599–1605. [Google Scholar] [CrossRef]
- Patrauchan, M.A.; Florizone, C.; Eapen, S.; Gomez-Gil, L.; Sethuraman, B.; Fukuda, M.; Davies, J.; Mohn, W.W.; Eltis, L.D. Roles of ring-hydroxylating dioxygenases in styrene and benzene catabolism in Rhodococcus jostii RHA1. J. Bacteriol. 2008, 190, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.; Lee, K.; Gibson, D. Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp strain NCIB 9816. J. Ind. Microbiol. 1996, 17, 438–457. [Google Scholar]
- Yen, K.-M.; Karl, M.R.; Blatt, L.M.; Simon, M.J.; Winter, R.B.; Fausset, P.R.; Lu, H.S.; Harcourt, A.A.; Chen, K.K. Cloning and characterization of a Pseudomonas mendocina KR1 gene cluster encoding toluene-4-monooxygenase. J. Bacteriol. 1991, 173, 5315–5327. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Mao, G.; Wang, Y.; Bartlam, M. Structural insights into diversity and n-alkane biodegradation mechanisms of alkane hydroxylases. Front. Microbiol. 2013, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.J.; Zhu, M.; Tso, L.; Nguyen, H.-H.T.; Yip, J.H.-K.; Chan, S.I. Regio-and stereoselectivity of particulate methane monooxygenase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc. 1997, 119, 9949–9955. [Google Scholar] [CrossRef]
- Smith, T.; Dalton, H. Biocatalysis by methane monooxygenase and its implications for the petroleum industry. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; Volume 151, pp. 177–192. [Google Scholar]
- Johnson, E.L.; Hyman, M.R. Propane and n-butane oxidation by Pseudomonas putida GPo1. Appl. Environ. Microbiol. 2006, 72, 950–952. [Google Scholar] [CrossRef]
- Li, L.; Liu, X.; Yang, W.; Xu, F.; Wang, W.; Feng, L.; Bartlam, M.; Wang, L.; Rao, Z. Crystal structure of long-chain alkane monooxygenase (LadA) in complex with coenzyme FMN: Unveiling the long-chain alkane hydroxylase. J. Mol. Biol. 2008, 376, 453–465. [Google Scholar] [CrossRef]
- van Beilen, J.B.; Wubbolts, M.G.; Witholt, B. Genetics of alkane oxidation by Pseudomonas oleovorans. Biodegradation 1994, 5, 161–174. [Google Scholar] [CrossRef]
- Stainthorpe, A.; Lees, V.; Salmond, G.P.; Dalton, H.; Murrell, J.C. The methane monooxygenase gene cluster of Methylococcus capsulatus (Bath). Gene 1990, 91, 27–34. [Google Scholar] [CrossRef]
- Chan, S.I.; Chen, K.H.-C.; Yu, S.S.-F.; Chen, C.-L.; Kuo, S.S.-J. Toward delineating the structure and function of the particulate methane monooxygenase from methanotrophic bacteria. Biochemistry 2004, 43, 4421–4430. [Google Scholar] [CrossRef]
- Tavormina, P.L.; Ussler, W., III; Joye, S.B.; Harrison, B.K.; Orphan, V.J. Distributions of putative aerobic methanotrophs in diverse pelagic marine environments. ISME J. 2010, 4, 700. [Google Scholar] [CrossRef] [PubMed]
- Meynet, P.; Head, I.M.; Werner, D.; Davenport, R.J. Re-evaluation of dioxygenase gene phylogeny for the development and validation of a quantitative assay for environmental aromatic hydrocarbon degraders. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.; Pieper, D. Genetics of biphenyl biodegradation and co-metabolism of PCBs. In Handbook of Hydrocarbon and Lipid Microbiology; Timmis, K.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 1179–1199. [Google Scholar]
- Iwai, S.; Chai, B.; Sul, W.J.; Cole, J.R.; Hashsham, S.A.; Tiedje, J.M. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 2010, 4, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Johnson, T.A.; Chai, B.; Hashsham, S.A.; Tiedje, J.M. Comparison of the specificities and efficacies of primers for aromatic dioxygenase gene analysis of environmental samples. Appl. Environ. Microbiol. 2011, 77, 3551–3557. [Google Scholar] [CrossRef] [PubMed]
- Grund, E.; Denecke, B.; Eichenlaub, R. Naphthalene degradation via salicylate and gentisate by Rhodococcus sp. strain B4. Appl. Environ. Microbiol. 1992, 58, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Di Gennaro, P.; Terreni, P.; Masi, G.; Botti, S.; De Ferra, F.; Bestetti, G. Identification and characterization of genes involved in naphthalene degradation in Rhodococcus opacus R7. Appl. Microbiol. Biotechnol. 2010, 87, 297–308. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Sanz, D.; Redondo-Nieto, M.; Martín, M.; Rivilla, R. Comparative Genomics of the Rhodococcus Genus Shows Wide Distribution of Biodegradation Traits. Microorganisms 2020, 8, 774. https://doi.org/10.3390/microorganisms8050774
Garrido-Sanz D, Redondo-Nieto M, Martín M, Rivilla R. Comparative Genomics of the Rhodococcus Genus Shows Wide Distribution of Biodegradation Traits. Microorganisms. 2020; 8(5):774. https://doi.org/10.3390/microorganisms8050774
Chicago/Turabian StyleGarrido-Sanz, Daniel, Miguel Redondo-Nieto, Marta Martín, and Rafael Rivilla. 2020. "Comparative Genomics of the Rhodococcus Genus Shows Wide Distribution of Biodegradation Traits" Microorganisms 8, no. 5: 774. https://doi.org/10.3390/microorganisms8050774
APA StyleGarrido-Sanz, D., Redondo-Nieto, M., Martín, M., & Rivilla, R. (2020). Comparative Genomics of the Rhodococcus Genus Shows Wide Distribution of Biodegradation Traits. Microorganisms, 8(5), 774. https://doi.org/10.3390/microorganisms8050774