A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion


Abstract
1. Introduction
2. Materials and Methods
2.1. Simulated Metagenomics Mock Data Sets
2.2. Tool Evaluation
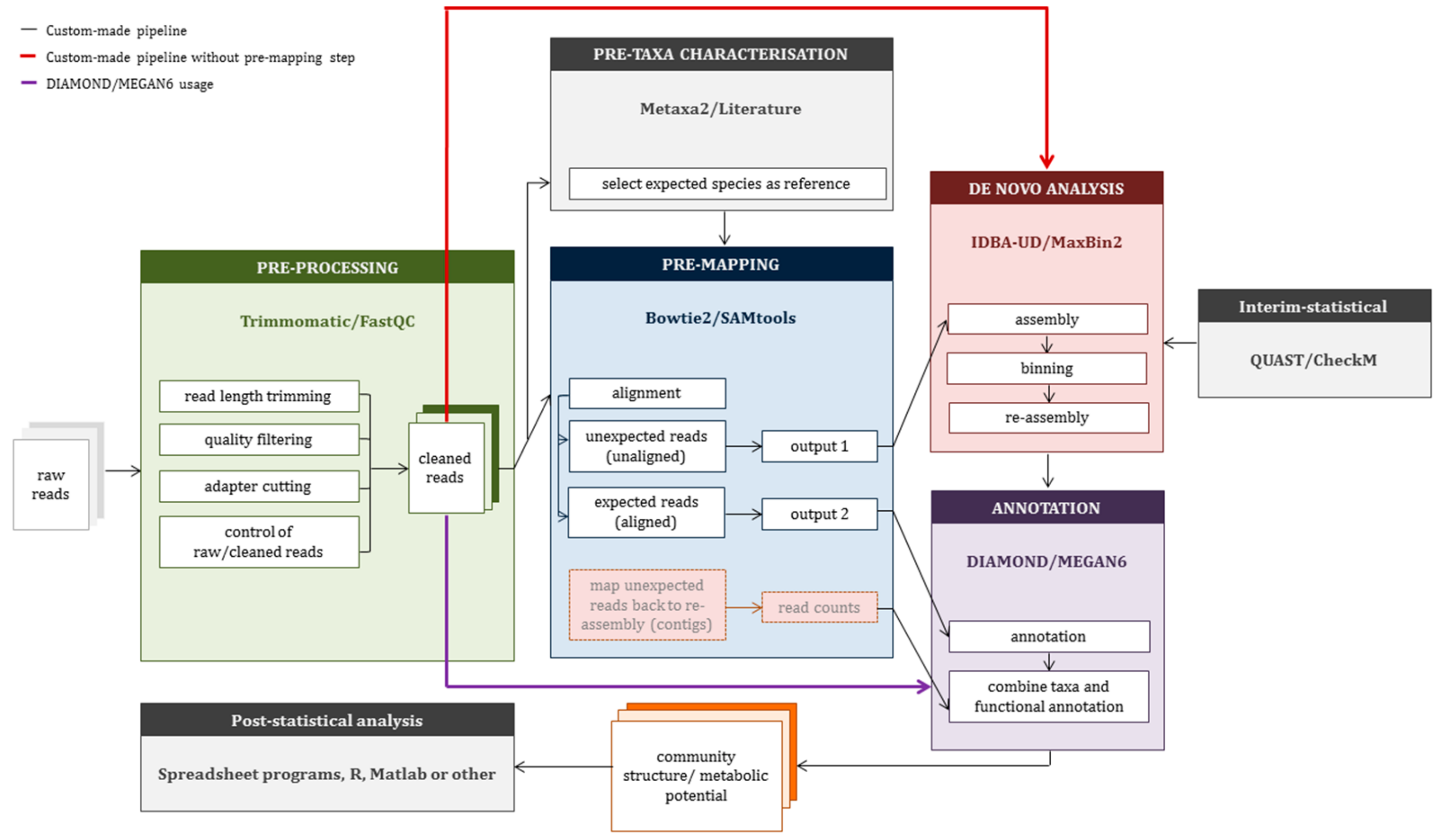
2.3. Construction of a Custom-Made Pipeline (CMP)
3. Results
3.1. Tool Selection
3.2. Application of Custom-Made Pipeline to Mock Data Sets
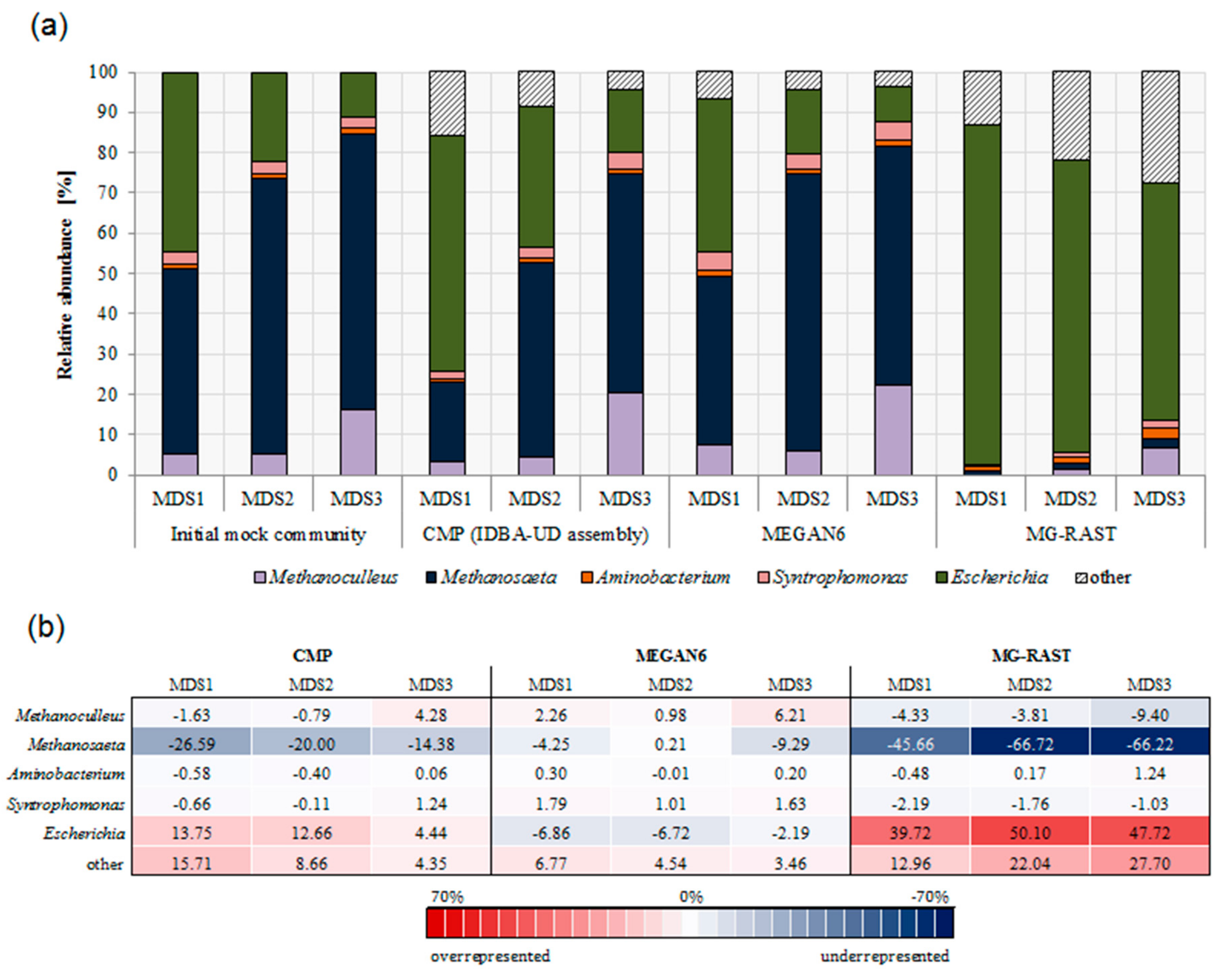
3.3. Comparison of Community Composition
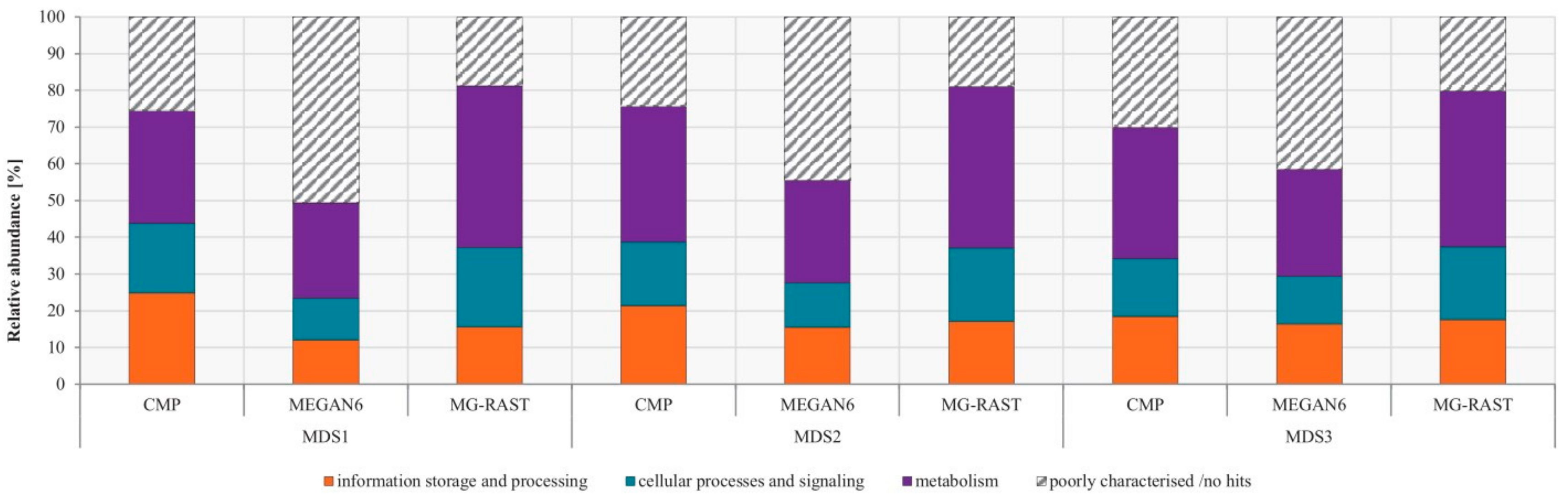
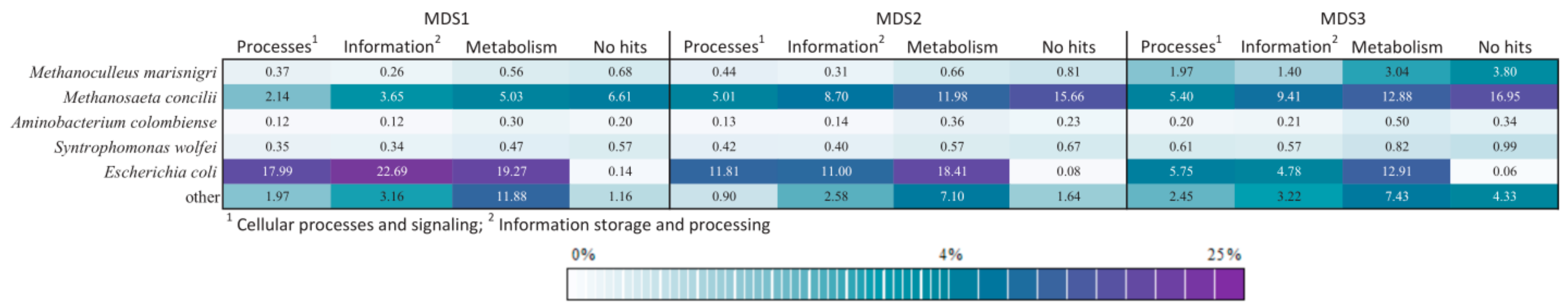
3.4. Comparison of Functional Potential
3.5. Advantage of Including Expected Species as Prior Knowledge in Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Copeland, A.; Lapidus, A.; Mavromatis, K.; Hugenholtz, P. A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. 2008, 72, 557–578. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Erratum: Corrigendum: Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2018, 36, 660-660. [Google Scholar] [CrossRef]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic assembly: Overview, challenges and applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. MetaVelvet: An extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next-generation sequencing data. Genomics 2010, 95, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Papudeshi, B.; Haggerty, J.M.; Doane, M.; Morris, M.M.; Walsh, K.; Beattie, D.T.; Pande, D.; Zaeri, P.; Silva, G.G.Z.; Thompson, F.; et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics 2017, 18, 915. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Kougias, P.G.; De Francisci, D.; Valle, G.; Angelidaki, I. Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol. Biofuels 2016, 9, 26. [Google Scholar] [CrossRef]
- Wu, Y.W.; Tang, Y.H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L.S.; Venter, C.; Remington, K.; Heidelberg, J.; Halpern, A.; Rusch, D.; Eisen, J.; Wu, D.; Paulsen, I.; et al. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Fosso, B.; Santamaria, M.; D’Antonio, M.; Lovero, D.; Corrado, G.; Vizza, E.; Passaro, N.; Garbuglia, A.R.; Capobianchi, M.R.; Crescenzi, M.; et al. MetaShot: An accurate workflow for taxon classification of host-associated microbiome from shotgun metagenomic data. Bioinformatics 2017, 33, 1730–1732. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2020, 48, D570–D578. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wilke, A.; Bischof, J.; Gerlach, W.; Glass, E.; Harrison, T.; Keegan, K.P.; Paczian, T.; Trimble, W.L.; Bagchi, S.; Grama, A.; et al. The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2016, 44, D590–D594. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- De Vrieze, J.; Raport, L.; Roume, H.; Vilchez-Vargas, R.; Jáuregui, R.; Pieper, D.H.; Boon, N. The full-scale anaerobic digestion microbiome is represented by specific marker populations. Water Res. 2016, 104, 101–110. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Jackman, S.D.; Vandervalk, B.P.; Mohamadi, H.; Chu, J.; Yeo, S.; Hammond, S.A.; Jahesh, G.; Khan, H.; Coombe, L.; Warren, R.L.; et al. ABySS 2. 0: Resource-Efficient Assembly of Large Genomes using a Bloom Filter Effect of Bloom Filter False Positive Rate. Genome Res. 2017, 27, 768–777. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA—A practical iterative De Bruijn graph De Novo assembler. In Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics); Springer: Berlin/Heidelberg, Germany, 2010; Volume 6044 LNBI, pp. 426–440. [Google Scholar]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- NCBI Genome Database. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 18 August 2017).
- Angly, F.E.; Willner, D.; Rohwer, F.; Hugenholtz, P.; Tyson, G.W. Grinder: A versatile amplicon and shotgun sequence simulator. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- BBMap. Available online: http://sourceforge.net/projects/bbmap/ (accessed on 30 September 2009).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S.; Adair, K.L.; Gardner, P.P. An evaluation of the accuracy and speed of metagenome analysis tools. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.; Krueger, F.; Seconds-Pichon, A.; Biggins, F.; Wingett, S. FastQC. A quality control tool for high throughput sequence data. Babraham Bioinformatics. Babraham Inst. 2015, 1, 1. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Hartmann, M.; Eriksson, K.M.; Pal, C.; Thorell, K.; Larsson, D.G.J.; Nilsson, R.H. metaxa2: Improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 2015, 15, 1403–1414. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, H.; Zhang, J.; Zhang, L.; Geng, A.; Liu, F.; Zhao, G.; Wang, S.; Zhou, Z.; Yan, X. Insight into dominant cellulolytic bacteria from two biogas digesters and their glycoside hydrolase genes. PLoS ONE 2015, 10, e0129921. [Google Scholar] [CrossRef]
- Stolze, Y.; Zakrzewski, M.; Maus, I.; Eikmeyer, F.; Jaenicke, S.; Rottmann, N.; Siebner, C.; Pühler, A.; Schlüter, A. Comparative metagenomics of biogas-producing microbial communities from production-scale biogas plants operating under wet or dry fermentation conditions. Biotechnol. Biofuels 2015, 8. [Google Scholar] [CrossRef]
- Goswami, R.; Chattopadhyay, P.; Shome, A.; Banerjee, S.N.; Chakraborty, A.K.; Mathew, A.K.; Chaudhury, S. An overview of physico-chemical mechanisms of biogas production by microbial communities: A step towards sustainable waste management. 3 Biotech 2016, 6, 72. [Google Scholar] [CrossRef]
- Nolla-Ardevol, V.; Strous, M.; Tegetmeyer, H.E. Anaerobic digestion of the microalga Spirulina at extreme alkaline conditions: Biogas production, metagenome and metatranscriptome. Front. Microbiol. 2015, 6, 597. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Strömberg, M.P.; Marth, G.T. Bamtools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- FastX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 8 January 2019).
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- Protein Accession to EggNOG Mapping File (MEGAN6). Available online: http://ab.inf.uni-tuebingen.de/data/software/megan6/download/acc2eggnog-Oct2016X.abin.zip (accessed on 29 November 2018).
- Protein Accession to NCBI-taxonomy Mapping File. Available online: http://ab.inf.uni-tuebingen.de/data/software/megan6/download/prot_acc2tax-ov2018×1.abin.zip (accessed on 29 November 2018).
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Segata, N.; Boernigen, D.; Tickle, T.L.; Morgan, X.C.; Garrett, W.S.; Huttenhower, C. Computational meta’omics for microbial community studies. Mol. Syst. Biol. 2013, 9, 666. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McCall, C.; Xagoraraki, I. Comparative study of sequence aligners for detecting antibiotic resistance in bacterial metagenomes. Lett. Appl. Microbiol. 2018, 66, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | NCBI Reference Sequence | Genome Size [bp] | Abundance [%] | ||
|---|---|---|---|---|---|
| MDS1 | MDS2 | MDS3 | |||
| Methanoculleus marisnigri | NC_009051.1 | 2,478,101 | 5.03 | 5.08 | 16.22 |
| Methanosaeta concilii1 | NC_015416.1 NC_015430.1 | 3,008,626 18,019 | 46.08 | 68.45 | 68.44 |
| Aminobacterium colombiense | NC_014011.1 | 1,980,592 | 1.35 | 1.35 | 1.34 |
| Syntrophomonas wolfei | NC_008346.1 | 2,936,195 | 2.80 | 2.80 | 2.80 |
| Escherichia coli | NC_000913.3 | 4,641,652 | 44.74 | 22.37 | 11.19 |
| Criteria | Description |
|---|---|
| Availability | Open source or commercial version as download or web service. |
| Support | Availability of: manual/readme, version update, help functions, and/or developer contact. |
| Flexibility | Flexible usage (variety of input/output data formats). |
| Run time | Expenditure of time: Operating time. |
| Usability | Ease of tool usage (installation, parameter settings, algorithm applicability). |
| Step | Feature | Description |
|---|---|---|
| Assembly | Number of contigs | Number of contigs generated |
| Largest contig | Length of the longest contig | |
| N50 | Median statistic whereby 50% of the entire assembly (in terms of number of bases) is contained in contigs longer or equal to the N50 value. Assemblies can only be compared if the assembly size is similar [14]. | |
| Total assembly length | Sum over all bases of all contigs generated | |
| Binning | Number of bins | Number of bins (clusters) |
| Completeness/contamination | CheckM compares completeness and contamination per bin depending on a defined number of marker genes for bacterial and archaeal genomes as a quality factor. |
| Criteria | MDS1 | MDS2 | MDS3 |
|---|---|---|---|
| Assembly | |||
| number contigs | 66 | 167 | 966 |
| largest contig [bp] | 327,353 | 145,780 | 16,676 |
| total assembly length [bp] | 4,564,668 | 4,520,232 | 2,975,790 |
| N50 [bp] | 133,300 | 47,255 | 3,611 |
| Reassembly | |||
| number contigs | 72 | 75 | 672 |
| largest contig [bp] | 327,353 | 317,670 | 27,558 |
| total assembly length [bp] | 4,567,420 | 4,378,346 | 3,381,373 |
| N50 [bp] | 132,848 | 113,839 | 5,862 |
| CMP with Pre-Mapping (Only Unexpected Reads) | CMP without Pre-Mapping(All Reads) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDS | Bins | MM | MC | AC | SW | EC | other | MM | MC | AC | SW | EC | other |
| 1 | nc | 0.00 | 0.00 | 0.00 | 0.00 | 90.30 | 9.70 | 16.25 | 3.48 | 19.33 | 59.35 | 0.20 | 1.39 |
| 1 | -- | -- | -- | -- | -- | -- | 0.00 | 2.22 | 0.00 | 0.00 | 88.25 | 9.53 | |
| 2 | -- | -- | -- | -- | -- | -- | 0.00 | 99.60 | 0.00 | 0.27 | 0.00 | 0.13 | |
| 2 | nc | 0.00 | 0.00 | 0.00 | 0.00 | 89.80 | 10.20 | 13.73 | 14.89 | 15.81 | 49.17 | 4.76 | 1.64 |
| 1 | 0.00 | 0.00 | 0.00 | 0.00 | 96.63 | 3.37 | 0.65 | 81.42 | 0.79 | 2.55 | 13.33 | 1.26 | |
| 2 | -- | -- | -- | -- | -- | -- | 0.79 | 50.57 | 0.95 | 3.10 | 40.27 | 4.32 | |
| 3 | nc | 0.00 | 0.00 | 0.00 | 0.00 | 89.21 | 10.79 | 0.96 | 6.52 | 13.19 | 39.82 | 35.36 | 4.16 |
| 1 | 0.00 | 0.00 | 0.00 | 0.00 | 94.38 | 5.62 | 3.26 | 82.57 | 1.05 | 3.47 | 8.85 | 0.80 | |
| 2 | 0.00 | 0.00 | 0.00 | 0.00 | 88.12 | 11.88 | 18.55 | 59.36 | 1.50 | 4.93 | 12.67 | 2.99 | |
| 3 | -- | -- | -- | -- | -- | -- | 3.46 | 45.54 | 1.15 | 3.77 | 41.53 | 4.55 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, D.; Popp, D.; Harms, H.; Centler, F. A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion. Microorganisms 2020, 8, 669. https://doi.org/10.3390/microorganisms8050669
Becker D, Popp D, Harms H, Centler F. A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion. Microorganisms. 2020; 8(5):669. https://doi.org/10.3390/microorganisms8050669
Chicago/Turabian StyleBecker, Daniela, Denny Popp, Hauke Harms, and Florian Centler. 2020. "A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion" Microorganisms 8, no. 5: 669. https://doi.org/10.3390/microorganisms8050669
APA StyleBecker, D., Popp, D., Harms, H., & Centler, F. (2020). A Modular Metagenomics Pipeline Allowing for the Inclusion of Prior Knowledge Using the Example of Anaerobic Digestion. Microorganisms, 8(5), 669. https://doi.org/10.3390/microorganisms8050669