Mycobacterium bovis: From Genotyping to Genome Sequencing

Abstract
1. Introduction
2. A Brief Background on MTBC Genomics
3. Traditional Genotyping Techniques of M. bovis
3.1. Restriction Endonuclease Analysis and Pulsed-Field Gel Electrophoresis
3.2. IS6110-RFLP
3.3. PGRS-RFLP
3.4. Spoligotyping
3.5. Variable Number Tandem Repeat (VNTR)
4. The Dawn of a New Era: WGS to Understand M. bovis Epidemiology and Ecology
4.1. Current WGS Workflow
Overview
5. Data Analyses Pipeline
5.1. Quality Assessment of Entry Data
5.2. Choice of Reference Genome for Read Mapping
5.3. Reads Mapping and Variant Calling
5.4. Within-Host Genetic Diversity and Its Impact on Variant Calling
5.5. Within-Host Genetic Diversity and Its Impact on Transmission Detection
5.6. Where to Go after Detection of Variants?
5.6.1. SNP-Counting Method
5.6.2. Whole-Genome Based Multi-Locus Sequencing Typing
5.6.3. Phylogenetic Approaches
6. Errors Arising from Indels and Repetitive Regions
7. Software to Define Spoligotyping and MIRU-VNTR Profiles Using WGS Data
8. Association of WGS with Epidemiological Data for Transmission Inference
9. Data Reporting in WGS Pipelines
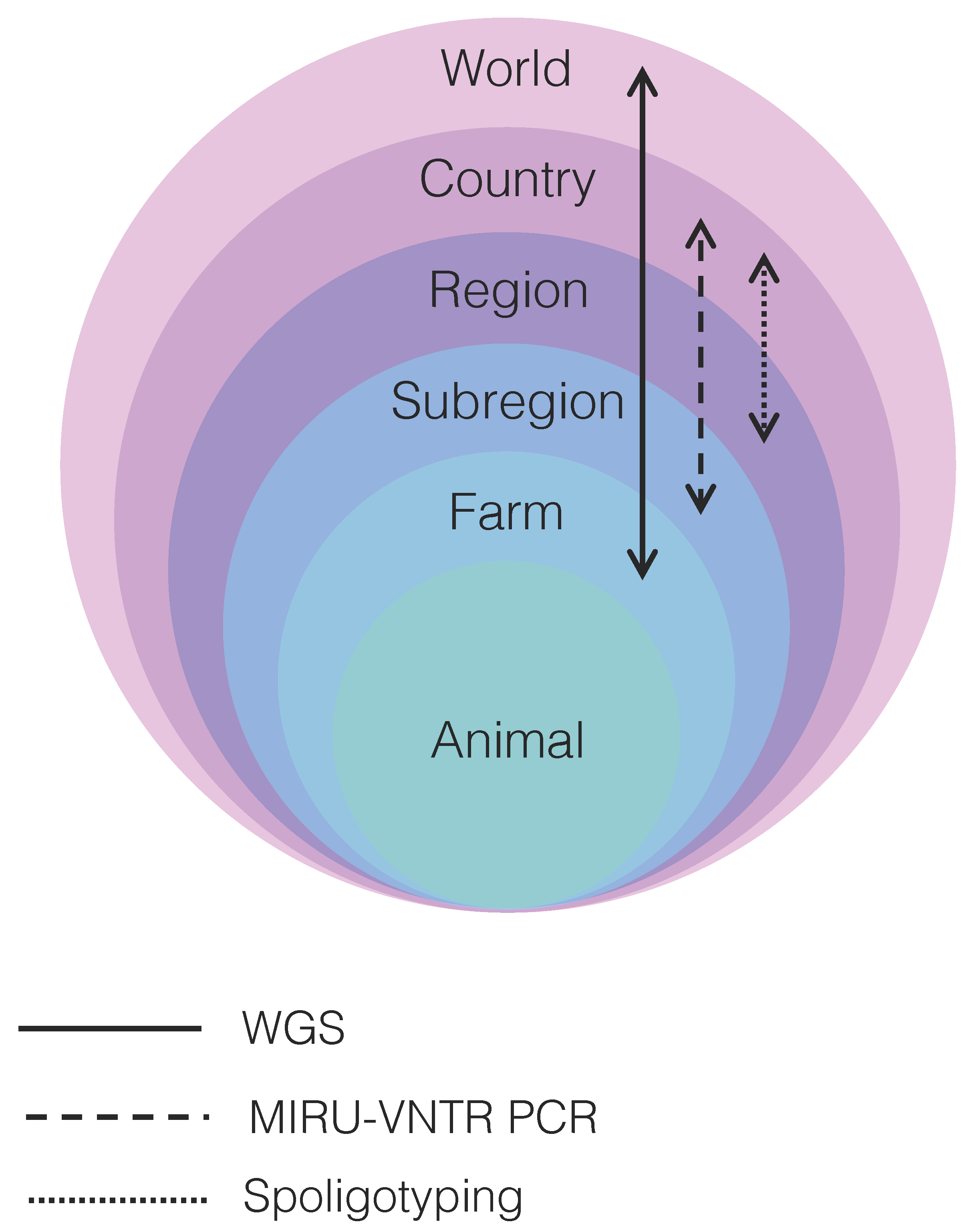
10. Resolution Power of WGS and Genotyping Techniques
11. WGS Provides New Insights into the Global Distribution of M. bovis Lineages
12. Other Pathogens Causing bTB
13. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galagan, J.E. Genomic insights into tuberculosis. Nat. Rev. Genet. 2014, 15, 307–320. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Tuberculosis Report 2019; World Health Organization: Geneve, Switzerland, 2019; ISBN 9789241565714. [Google Scholar]
- Cousing, S. End TB Strategy; WHO: Geneva, Switzerland, 2018; pp. 82–83. [Google Scholar]
- Ayele, W.Y.; Neill, S.D.; Zinsstag, J.; Weiss, M.G.; Pavlik, I. Bovine tuberculosis: An old disease but a new threat to Africa. Int. J. Tuberc. Lung Dis. 2004, 8, 924–937. [Google Scholar]
- Godfray, H.C.J.; Donnelly, C.A.; Kao, R.R.; Macdonald, D.W.; McDonald, R.A.; Petrokofsky, G.; Wood, J.L.N.; Woodroffe, R.; Young, D.B.; McLean, A.R. A restatement of the natural science evidence base relevant to the control of bovine tuberculosis in Great Britain. Proc. R. Soc. B Biol. Sci. 2013, 280, 20131634. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.S.; Sweeney, S.J. Mycobacterium bovis (bovine tuberculosis) infection in North American wildlife: Current status and opportunities for mitigation of risks of further infection in wildlife populations. Epidemiol. Infect. 2013, 141, 1357–1370. [Google Scholar] [CrossRef]
- Nugent, G.; Buddle, B.M.; Knowles, G. Epidemiology and control of Mycobacterium bovis infection in brushtail possums (Trichosurus vulpecula), the primary wildlife host of bovine tuberculosis in New Zealand. N. Z. Vet. J. 2015, 63, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Nugent, G.; Gortazar, C.; Knowles, G. The epidemiology of Mycobacterium bovis in wild deer and feral pigs and their roles in the establishment and spread of bovine tuberculosis in New Zealand wildlife. N. Z. Vet. J. 2015, 63, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.V. Mycobacterium bovis: Characteristics of Wildlife Reservoir Hosts. Transbound. Emerg. Dis. 2013, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Kantor, I.N.; Ritacco, V. An update on bovine tuberculosis programmes in Latin American and Caribbean countries. Vet. Microbiol. 2006, 112, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Olea-Popelka, F.; Muwonge, A.; Perera, A.; Dean, A.S.; Mumford, E.; Erlacher-Vindel, E.; Forcella, S.; Silk, B.J.; Ditiu, L.; El Idrissi, A.; et al. Zoonotic tuberculosis in human beings caused by Mycobacterium bovis—A call for action. Lancet Infect. Dis. 2017, 17, e21–e25. [Google Scholar] [CrossRef]
- Cosivi, O.; Grange, J.M.; Daborn, C.J.; Raviglione, M.C.; Fujikura, T.; Cousins, D.; Robinson, R.A.; Huchzermeyer, H.F.A.K.; de Kantor, I.; Meslin, F.X. Zoonotic tuberculosis due to Mycobacterium bovis in developing countries. Emerg. Infect. Dis. 1998, 4, 59–70. [Google Scholar] [CrossRef]
- Loiseau, C.; Brites, D.; Moser, I.; Coll, F.; Pourcel, C.; Robbe-Austerman, S.; Escuyer, V.; Musser, K.A.; Peacock, S.J.; Feuerriegel, S.; et al. Revised interpretation of the Hain Lifescience Genotype MTBC to differentiate Mycobacterium canettii and members of the Mycobacterium tuberculosis complex. Antimicrob. Agents Chemother. 2019, 63, 1–13. [Google Scholar] [CrossRef]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Feldman, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar] [PubMed]
- Dürr, S.; Müller, B.; Alonso, S.; Hattendorf, J.; Laisse, C.J.M.; van Helden, P.D.; Zinsstag, J. Differences in primary sites of infection between zoonotic and human tuberculosis: Results from a worldwide systematic review. PLoS Negl. Trop. Dis. 2013, 7, e2399. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO); Food and Agriculture Organization of the United Nations (FAO); World Organisation for Animal Health (OIE). Roadmap for Zoonotic Tuberculosis; World Health Organization: Geneva, Switzerland, 2017; ISBN 9789241513043. [Google Scholar]
- OIE. Bovine tuberculosis. Gen. Dis. Inf. Sheets 2011, 1–6. [Google Scholar]
- Anderson, D.P.; Ramsey, D.S.L.; de Lisle, G.W.; Bosson, M.; Cross, M.L.; Nugent, G. Development of integrated surveillance systems for the management of tuberculosis in New Zealand wildlife. N. Z. Vet. J. 2015, 63, 89–97. [Google Scholar] [CrossRef]
- Skuce, R.A.; Mallon, T.R.; McCormick, C.M.; McBride, S.H.; Clarke, G.; Thompson, A.; Couzens, C.; Gordon, A.W.; McDowell, S.W.J. Mycobacterium bovis genotypes in Northern Ireland: Herd-level surveillance (2003 to 2008). Vet. Rec. 2010, 167, 684–689. [Google Scholar] [CrossRef]
- Orloski, K.; Robbe-austerman, S.; Stuber, T.; Hench, B.; Schoenbaum, M. Whole genome sequencing of Mycobacterium bovis isolated from livestock in the United States, 1989–2018. Front. Vet. Sci. 2018, 5, 1–23. [Google Scholar] [CrossRef]
- Réveillaud, É.; Desvaux, S.; Boschiroli, M.-L.; Hars, J.; Faure, É.; Fediaevsky, A.; Cavalerie, L.; Chevalier, F.; Jabert, P.; Poliak, S.; et al. Infection of wildlife by Mycobacterium bovis in France assessment through a national surveillance system, Sylvatub. Front. Vet. Sci. 2018, 5, 262. [Google Scholar] [CrossRef]
- Ramos, D.F.; Tavares, L.; da Silva, P.E.A.; Dellagostin, O.A. Molecular typing of Mycobacterium bovis isolates: A review. Braz. J. Microbiol. 2014, 45, 365–372. [Google Scholar] [CrossRef]
- El-Sayed, A.; El-Shannat, S.; Kamel, M.; Castañeda-Vazquez, M.A.; Castañeda-Vazquez, H. Molecular Epidemiology of Mycobacterium bovis in Humans and Cattle. Zoonoses Public Health 2016, 63, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M. Advances in molecular diagnostics for Mycobacterium bovis. Vet. Microbiol. 2011, 151, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.R.; Price-Carter, M.; Robbe-Austerman, S. Use of genomics to track bovine tuberculosis transmission. OIE Rev. Sci. Tech. 2016, 35, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Durr, P.A.; Hewinson, R.G.; Clifton-Hadley, R.S. Molecular epidemiology of bovine tuberculosis. I. Mycobacterium bovis genotyping. Rev. Sci. Tech. 2000, 19, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Haddad, N.; Masselot, M.; Durand, B. Molecular differentiation of Mycobacterium bovis isolates. Review of main techniques and applications. Res. Vet. Sci. 2004, 76, 1–18. [Google Scholar] [CrossRef]
- Merker, M.; Kohl, T.A.; Niemann, S.; Supply, P. The evolution of strain typing in the Mycobacterium tuberculosis complex. Adv. Exp. Med. Biol. 2017, 1019, 79–93. [Google Scholar]
- Tsao, K.; Robbe-Austerman, S.; Miller, R.S.; Portacci, K.; Grear, D.A.; Webb, C. Sources of bovine tuberculosis in the United States. Infect. Genet. Evol. 2014, 28, 137–143. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef]
- Bryant, J.; Chewapreecha, C.; Bentley, S.D. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 2012, 7, 1283–1296. [Google Scholar] [CrossRef]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef]
- Meehan, C.J.; Goig, G.A.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Microbiol. 2019, 17, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Crisan, A.; Gardy, J.L.; Munzner, T. A systematic method for surveying data visualizations and a resulting genomic epidemiology visualization typology: GEViT. Bioinformatics 2019, 35, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Belak, S.; Karlsson, O.E.; Leijon, M.; Granberg, F. High-throughput sequencing in veterinary infection biology and diagnostics. Rev. Sci. Tech. l’OIE 2013, 32, 893–915. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.W.; Strain, E.; Melka, D.; Bunning, K.; Musser, S.M.; Brown, E.W.; Timme, R. Practical value of food pathogen traceability through building a whole-genome sequencing network and database. J. Clin. Microbiol. 2016, 54, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- OIE (World Organisation for Animal Health). Chapter 1.1.7. Standards for High. Throughput Sequencing, Bioinformatics and Computational Genomics, 8th ed.; OIE (World Organisation for Animal Health): Paris, France, 2018. [Google Scholar]
- Van Bomr, S.; Wang, J.; Granberg, F.; Colling, A. Next-generation sequencing workflows in veterinary infection biology: Towards validation and quality assurance. Rev. Sci. Tech. l’OIE 2016, 35, 67–81. [Google Scholar]
- World Health Organization (WHO). Whole Genome Sequencing for Foodborne Disease Surveillance: Landscape Paper; World Health Organization (WHO): Geneva, Switzerland, 2018; ISBN 9789241513869. [Google Scholar]
- World Health Organization (WHO). The Use of Next-Generation Sequencing Technologies for the Detection of Mutations Associated with Drug Resistance in Mycobacterium tuberculosis Complex.: Technical Guide; World Health Organization (WHO): Geneva, Switzerland, 2018. [Google Scholar]
- Tagliani, E.; Cirillo, D.M.; Ködmön, C.; van der Werf, M.J. EUSeqMyTB to set standards and build capacity for whole genome sequencing for tuberculosis in the EU. Lancet Infect. Dis. 2018, 18, 377. [Google Scholar] [CrossRef]
- Glaser, L.; Carstensen, M.; Shaw, S.; Robbe-Austerman, S.; Wunschmann, A.; Grear, D.; Stuber, T.; Thomsen, B. Descriptive epidemiology and whole genome sequencing analysis for an outbreak of bovine tuberculosis in beef cattle and white-tailed deer in northwestern Minnesota. PLoS ONE 2016, 11, e0145735. [Google Scholar] [CrossRef]
- Broeckl, S.; Krebs, S.; Varadharajan, A.; Straubinger, R.K.; Blum, H.; Buettner, M. Investigation of intra-herd spread of Mycobacterium caprae in cattle by generation and use of a whole-genome sequence. Vet. Res. Commun. 2017, 41, 113–128. [Google Scholar] [CrossRef]
- Zimpel, C.K.; Patané, J.S.L.; Guedes, A.C.P.; Souza, R.F.; Pereira-Silva, T.T.; Soler Camargo, N.C.; de Souza Filho, A.F.; Ikuta, C.Y.; Soares Ferreira Neto, J.; Setubal, J.C.; et al. Global distribution and evolution of Mycobacterium bovis. Front. Microbiol. 2020, 11, 1–19. [Google Scholar]
- Crispell, J.; Zadoks, R.N.; Harris, S.R.; Paterson, B.; Collins, D.M.; De-Lisle, G.W.; Livingstone, P.; Neill, M.A.; Biek, R.; Lycett, S.J.; et al. Using whole genome sequencing to investigate transmission in a multi-host system: Bovine tuberculosis in New Zealand. BMC Genom. 2017, 18, 180. [Google Scholar] [CrossRef]
- Kohl, T.A.; Utpatel, C.; Niemann, S.; Moser, I. Mycobacterium bovis persistence in two different captive wild animal populations in Germany: A longitudinal molecular epidemiological study revealing pathogen transmission by whole-genome sequencing. J. Clin. Microbiol. 2018, 56, 1–9. [Google Scholar] [CrossRef]
- Ghebremariam, M.K.; Hlokwe, T.; Rutten, V.P.M.G.; Allepuz, A.; Cadmus, S.; Muwonge, A.; Robbe-Austerman, S.; Michel, A.L. Genetic profiling of Mycobacterium bovis strains from slaughtered cattle in Eritrea. PLoS Negl. Trop. Dis. 2018, 12, e0006406. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, H.F.M.; Spalink, D.; Amer, A.; Steinberg, H.; Hashish, E.A.; Nasr, E.A.; Talaat, A.M. Genomic Polymorphism Associated with the Emergence of Virulent Isolates of Mycobacterium bovis in the Nile Delta. Sci. Rep. 2019, 9, 11657. [Google Scholar] [CrossRef] [PubMed]
- Salvador, L.C.M.; O’Brien, D.J.; Cosgrove, M.K.; Stuber, T.P.; Schooley, A.M.; Crispell, J.; Church, S.V.; Gröhn, Y.T.; Robbe-Austerman, S.; Kao, R.R. Disease management at the wildlife-livestock interface: Using whole-genome sequencing to study the role of elk in Mycobacterium bovis transmission in Michigan, USA. Mol. Ecol. 2019, 28, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Crispell, J.; Benton, C.H.; Balaz, D.; De Maio, N.; Akhmetova, A.; Allen, A.; Biek, R.; Presho, E.L.; Dale, J.; Hewinson, G.; et al. Combining genomics and epidemiology to analyse bi-directional transmission of Mycobacterium bovis in a multi-host system. Elife 2019, 8, e45833. [Google Scholar] [CrossRef]
- Gordon, S. Strain Variation in the Mycobacterium Tuberculosis Complex: Its Role in Biology, Epidemiology and Control; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1019. [Google Scholar]
- Supply, P.; Warren, R.M.; Bañuls, A.-L.; Lesjean, S.; Van Der Spuy, G.D.; Lewis, L.-A.; Tibayrenc, M.; Van Helden, P.D.; Locht, C. Linkage disequilibrium between minisatellite loci supports clonal evolution of Mycobacterium tuberculosis in a high tuberculosis incidence area. Mol. Microbiol. 2003, 47, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Gagneux, S.; Small, P.M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 2007, 7, 328–337. [Google Scholar] [CrossRef]
- Hirsh, A.E.; Tsolaki, A.G.; DeRiemer, K.; Feldman, M.W.; Small, P.M. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc. Natl. Acad. Sci. USA 2004, 101, 4871–4876. [Google Scholar] [CrossRef]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 2013, 45, 1176–1182. [Google Scholar] [CrossRef]
- Bos, K.I.; Harkins, K.M.; Herbig, A.; Coscolla, M.; Weber, N.; Comas, I.; Forrest, S.A.; Bryant, J.M.; Harris, S.R.; Schuenemann, V.J.; et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 2014, 514, 494–497. [Google Scholar] [CrossRef]
- Brosch, R.; Gordon, S.V.; Marmiesse, M.; Brodin, P.; Buchrieser, C.; Eiglmeier, K.; Garnier, T.; Gutierrez, C.; Hewinson, G.; Kremer, K.; et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. USA 2002, 99, 3684–3689. [Google Scholar] [CrossRef]
- Gordon, S.V.; Brosch, R.; Billault, A.; Garnier, T.; Eiglmeier, K.; Cole, S.T. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol. Microbiol. 1999, 32, 643–655. [Google Scholar] [CrossRef]
- Brosch, R.; Gordon, S.V.; Billault, A.; Garnier, T.; Eiglmeier, K.; Soravito, C.; Barrell, B.G.; Cole, S.T. Use of a Mycobacterium tuberculosis H37Rv bacterial artificial chromosome library for genome mapping, sequencing, and comparative genomics. Infect. Immun. 1998, 66, 2221–2229. [Google Scholar] [CrossRef]
- Philipp, W.J.; Nair, S.; Guglielmi, G.; Lagrande, M.; Gicquelv, B.; Cole, S.T. Physical mapping of Mycobacterium bovis BCG Pasteur reveals differences from the genome map of Mycobacterium tuberculosis H37Rv and from M. bovis. Microbiology 2019, 142, 3135–3145. [Google Scholar] [CrossRef]
- Warren, R.M.; Van Pittius, N.C.G.; Barnard, M.; Hesseling, A.; Engelke, E.; De Kock, M.; Gutierrez, M.C.; Chege, G.K.; Victor, T.C.; Hoal, E.G.; et al. Differentiation of Mycobacterium tuberculosis complex by PCR amplification of genomic regions of difference. Int. J. Tuberc. Lung Dis. 2006, 10, 818–822. [Google Scholar]
- Karlson, A.G. Mycobacterium bovis nom.nov. Int. J. Syst. Bacteriol. 1970, 20, 273–282. [Google Scholar] [CrossRef]
- Goodfellow, M.; Kämpfer, P.; Busse, H.J.; Trujillo, M.E.; Suzuki, K.I.; Ludwig, W.W. Genus I. Streptomyces. In Bergey’s Manual of Systematic Bacteriology; Springer: New York, NY, USA, 2012; ISBN 978-0-387-95043-3. [Google Scholar]
- Wayne, L.; Kubica, G. The Mycobacteria: A Sourcebook; Marcel Dekker Inc.: New York, NY, USA, 1984; pp. 1436–1457. [Google Scholar]
- Shinnick, T.M.; Good, R.C. Mycobacterial taxonomy. Eur. J. Clin. Microbiol. Infect. Dis. 1994, 13, 884–901. [Google Scholar] [CrossRef]
- Baess, I. Deoxyribonucleic acid relatedness among species of slowly-growing mycobacteria. Acta Pathol. Microbiol. Scand. B 1979, 87, 221–226. [Google Scholar] [CrossRef]
- Smith, N.; Kremer, K.; Inwald, J.; Dale, J.; Driscoll, J.; Gordon, S.; van Soolingen, D.; Hewinson, R.; Smith, J. Ecotypes of the Mycobacterium tuberculosis complex. J. Theor. Biol. 2006, 239, 220–225. [Google Scholar] [CrossRef]
- Riojas, M.A.; McGough, K.J.; Rider-Riojas, C.J.; Rastogi, N.; Hazbón, M.H. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycob. Int. J. Syst. Evol. Microbiol. 2018, 68, 324–332. [Google Scholar] [CrossRef]
- Tyler, A.D.; Christianson, S.; Knox, N.C.; Mabon, P.; Wolfe, J.; Van Domselaar, G.; Graham, M.R.; Sharma, M.K. Comparison of sample preparation methods used for the next-generation sequencing of Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0148676. [Google Scholar] [CrossRef] [PubMed]
- Delogu, G.; Brennan, M.J.; Manganelli, R. PE and PPE genes: A tale of conservation and diversity. Adv. Exp. Med. Biol. 2017, 1019, 191–207. [Google Scholar] [PubMed]
- Tørresen, O.K.; Star, B.; Mier, P.; Andrade-navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, V.; Promponas, V.J.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef]
- Collins, D.M.; de Lisle, G.W. DNA Restriction Endonuclease Analysis of Mycobacterium tuberculosis and Mycobacterium bovis. BCG 1984, 130, 1019–1021. [Google Scholar] [CrossRef][Green Version]
- Collins, D.M.; De Lisle, G.W. DNA Restriction Endonuclease Analysis of Mycobacterium bovis and other members of the Tuberculosis complex. Microbiology 1985, 21, 526–564. [Google Scholar] [CrossRef]
- Collins, D.M.; De Lisle, G.W.; Gabric, D.M. Geographic distribution of restriction types of Mycobacterium bovis isolates from brush-tailed possums ( Trichosurus vulpecula) in New Zealand. J. Hyg. 1986, 96, 431–438. [Google Scholar] [CrossRef]
- Collins, D.M.; de Lisle, G.W.; Collins, J.D. DNA restriction fragment typing of Mycobacterium bovis isolates from cattle and badgers in Ireland. Vet. Rec. 1994, 134, 681–682. [Google Scholar] [CrossRef]
- Collins, D.M.; Gabric, D.M.; de Lisle, G.W. Typing of Mycobacterium bovis isolates from cattle and other animals in the same locality. N. Z. Vet. J. 1988, 36, 45–46. [Google Scholar] [CrossRef]
- Collins, D.M.; Erasmuson, S.K.; Stephens, D.M.; Yates, G.F.; De Lisle, G.W. DNA fingerprinting of Mycobacterium bovis strains by restriction fragment analysis and hybridization with insertion elements IS1081 and IS6110. J. Clin. Microbiol. 1993, 31, 1143–1147. [Google Scholar] [CrossRef]
- Zhang, Y.; Mazurek, G.H.; Cave, M.D.; Eisenach, K.D.; Pang, Y.; Murphy, D.T.; Wallace, R.J. DNA polymorphisms in strains of Mycobacterium tuberculosis analyzed by pulsed-field gel electrophoresis: A tool for epidemiology. J. Clin. Microbiol. 1992, 30, 1551–1556. [Google Scholar] [CrossRef]
- Hughes, V.M.; Stevenson, K.; Sharp, J.M. Improved preparation of high molecular weight DNA for pulsed-field gel electrophoresis from mycobacteria. J. Microbiol. Methods 2001, 44, 209–215. [Google Scholar] [CrossRef]
- Ghodousi, A.; Arash, G.; Vatani, S.; Darban-Sarokhalil, D.; Omrani, M.; Fooladi, A.; Khosaravi, A.; Feizabadi, M.M. Development of a new DNA extraction protocol for PFGE typing of Mycobacterium tuberculosis complex. Iran. J. Microbiol. 2012, 4, 44–46. [Google Scholar] [PubMed]
- Choe, Y.-K.; Huh, Y.-J.; Park, J.-H.; Kim, J.-R.; Park, J.-S.; Song, J.-C.; Ko, J.-H.; Lee, Y.-C.; Nashiru, O.; Kim, J.-K.; et al. Improved Isolation of Genomic DNA from Mycobacteria in Agarose Plugs by Rapid Lysis with a Combination of N-Acetylglucosaminidase and Lysozyme. Biotechniques 1996, 20, 547–552. [Google Scholar]
- Ravansalar, H.; Tadayon, K.; Ghazvini, K. Molecular typing methods used in studies of Mycobacterium tuberculosis in Iran: A systematic review. Iran. J. Microbiol. 2016, 8, 338–346. [Google Scholar]
- Jeon, S.; Lim, N.; Park, S.; Park, M.; Kim, S. Comparison of PFGE, IS6110-RFLP, and 24-Locus MIRU-VNTR for molecular epidemiologic typing of Mycobacterium tuberculosis isolates with known epidemic connections. J. Microbiol. Biotechnol. 2018, 28, 338–346. [Google Scholar] [CrossRef]
- Suffys, P.N.; De Araujo, M.E.I.; Degrave, W.M. The changing face of the epidemiology of tuberculosis due to molecular strain typing: A review. Mem. Inst. Oswaldo Cruz 1997, 92, 297–316. [Google Scholar] [CrossRef]
- Njanpop-Lafourcade, B.M.; Inwald, J.; Ostyn, A.; Durand, B.; Hughes, S.; Thorel, M.F.; Hewinson, G.; Haddad, N. Molecular typing of Mycobacterium bovis isolates from Cameroon. J. Clin. Microbiol. 2001, 39, 222–227. [Google Scholar] [CrossRef]
- Feizabadi, M.; Robertson, I.; Edwards, R.; Cousins, D.V.; Hampson, D. Genetic differentiation of Australian isolates of Mycobacterium tuberculosis by pulsed-field gel electrophoresis. J. Med. Microbiol. 1997, 46, 501–505. [Google Scholar] [CrossRef][Green Version]
- Hughes, V.M.; Skuce, R.; Doig, C.; Stevenson, K.; Sharp, J.M.; Watt, B. Analysis of multidrug-resistant Mycobacterium bovis from three clinical samples from Scotland. Int. J. Tuberc. Lung Dis. 2003, 7, 1191–1198. [Google Scholar]
- Thierry, D.; Brisson, N.A.; Vincent, L.F.; Nguyen, S.; Guesdon, J.L.; Gicquel, B. Characterization of Mycobacterium tuberculosis insertion sequence, IS6110, and its application in diagnosis. J. Clin. Microbiol. 1999, 28, 2668–2673. [Google Scholar] [CrossRef]
- Cave, M.D.; Eisenach, K.D.; McDermott, P.F.; Bates, J.H.; Crawford, J.T. IS6110: Conservation of sequence in the Mycobacterium tuberculosis complex and its utilization in DNA fingerprinting. Mol. Cell. Probes 1991, 5, 73–80. [Google Scholar] [CrossRef]
- Gonzalo-Asensio, J.; Pérez, I.; Aguiló, N.; Uranga, S.; Picó, A.; Lampreave, C.; Cebollada, A.; Otal, I.; Samper, S.; Martín, C. New insights into the transposition mechanisms of IS6110 and its dynamic distribution between Mycobacterium tuberculosis Complex lineages. PLoS Genet. 2018, 14, e1007282. [Google Scholar] [CrossRef]
- Thierry, D.; Cave, M.D.; Eisenach, K.D.; Crawford, J.T.; Bates, J.H.; Gicquel, B.; Guesdon, J.L.; Froides, S.; Microbiologique, D.G.; Pasteur, I. IS6110, an IS-like element of Mycobacterium tuberculosis. Nucleic Acids Res. 1990, 18, 6110. [Google Scholar] [CrossRef]
- Hermans, P.W.M.; Van Soolingen, D.; Dale, J.W.; Schuitema, A.R.J.; McAdam, R.A.; Catty, D.; Van Embden, J.D.A. Insertion element IS986 from Mycobacterium tuberculosis: A useful tool for diagnosis and epidemiology of tuberculosis. J. Clin. Microbiol. 1990, 28, 2051–2058. [Google Scholar] [CrossRef]
- Otal, I.; Martin, C.; Vincent-Levy-Frebault, V.; Thierry, D.; Gicquel, B. Restriction fragment length polymorphism analysis using IS6110 as an epidemiological marker in tuberculosis. J. Clin. Microbiol. 1991, 29, 1252–1254. [Google Scholar] [CrossRef]
- van Embden, J.D.; Cave, M.D.; Crawford, J.T.; Dale, J.W.; Eisenach, K.D.; Gicquel, B.; Hermans, P.; Martin, C.; McAdam, R.; Shinnick, T.M. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: Recommendations for a standardized methodology. J. Clin. Microbiol. 1993, 31, 406–409. [Google Scholar] [CrossRef]
- Warren, R.M.; Van Der Spuy, G.D.; Richardson, M.; Beyers, N.; Borgdorff, M.W.; Behr, M.A.; Helden, P.D. Van Calculation of the stability of the IS6110 banding pattern in patients with persistent Mycobacterium tuberculosis disease. J. Clin. Microbiol. 2002, 40, 1705–1708. [Google Scholar] [CrossRef]
- Van Soolingen, D.; Hermans, P.W.M.; De Haas, P.E.W.; Soll, D.R.; Van Embden, J.D.A. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: Evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin. Microbiol. 1991, 29, 2578–2586. [Google Scholar] [CrossRef]
- Cousins, D.V.; Williams, S.N.; Ross, B.C.; Ellis, T.M. Use of a repetitive element isolated from Mycobacterium tuberculosis in hybridization studies with Mycobacterium bovis: A new tool for epidemiological studies of bovine tuberculosis. Vet. Microbiol. 1993, 37, 1–17. [Google Scholar] [CrossRef]
- Ross, B.C.; Raios, K.; Jackson, K.; Sievers, A.; Dwyer, B. Differentiation of Mycobacterium tuberculosis strains by use of a nonradioactive Southern Blot hybridization method. J. Infect. Dis. 1991, 163, 904–907. [Google Scholar] [CrossRef]
- Ross, B.C.; Raios, K.; Jackson, K.; Dwyer, B. Molecular cloning of a highly repeated DNA element from Mycobacterium tuberculosis and its use as an epidemiological tool. J. Clin. Microbiol. 1992, 30, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Van Soolingen, D.; De Haas, P.E.W.; Hermans, P.W.M.; Groenen, P.M.A.; Van Embden, J.D.A. Comparison of various repetitive DNA elements as genetic markers for strain differentiation and epidemiology of Mycobacterium tuberculosis. J. Clin. Microbiol. 1993, 31, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Ijaz, K.; Bates, J.H.; Eisenach, K.D.; Cave, M.D. Spoligotyping and polymorphic GC-rich repetitive sequence fingerprinting of Mycobacterium tuberculosis strains having few copies of IS6110. J. Clin. Microbiol. 2000, 38, 3572–3576. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-Mediated Adaptive Immune Systems in Bacteria and Archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Hermans, P.W.M.; Van Soolingen, D.; Bik, E.M.; De Haas, P.E.W.; Dale, J.W.; Van Embden, J.D.A. Insertion element IS987 from Mycobacterium bovis BCG is located in a hot-spot integration region for insertion elements in Mycobacterium tuberculosis complex strains. Infect. Immun. 1991, 59, 2695–2705. [Google Scholar] [CrossRef]
- Groenen, P.M.A.; Bunschoten, A.E.; van Soolingen, D.; Errtbden, J.D.A. Nature of DNA polymorphism in the direct repeat cluster of Mycobacterium tuberculosis; application for strain differentiation by a novel typing method. Mol. Microbiol. 1993, 10, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Kamerbeek, J.; Schouls, L.E.O.; Kolk, A.; Kuijper, S.; Bunschoten, A.; Molhuizen, H.; Shaw, R.; Goyal, M. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997, 35, 907–914. [Google Scholar] [CrossRef]
- Aranaz, A.; Liébana, E.; Mateos, A.; Dominguez, L.; Vidal, D.; Domingo, M.; Gonzolez, O.; Rodriguez-Ferri, E.F.; Bunschoten, A.E.; Van Embden, J.D.A.; et al. Spacer oligonucleotide typing of Mycobacterium bovis strains from cattle and other animals: A tool for studying epidemiology of tuberculosis. J. Clin. Microbiol. 1996, 34, 2734–2740. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zanden, A.G.M.; Hoentjen, A.H.; Heilmann, F.G.C.; Weltevreden, E.F.; Schouls, L.M.; Van Embden, J.D.A. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis complex in paraffin wax embedded tissues and in stained microscopic preparations. J. Clin. Pathol. Mol. Pathol. 1998, 51, 209–214. [Google Scholar] [CrossRef]
- Cowan, L.S.; Diem, L.; Brake, M.C.; Crawford, J.T. Transfer of a Mycobacterium tuberculosis genotyping method, Spoligotyping, from a reverse line-blot hybridization, membrane-based assay to the Luminex multianalyte profiling system. J. Clin. Microbiol. 2004, 42, 474–477. [Google Scholar] [CrossRef]
- Ocheretina, O.; Merveille, Y.M.; Mabou, M.M.; Escuyer, V.E.; Dunbar, S.A.; Johnson, W.D.; Pape, J.W.; Fitzgerald, D.W. Use of Luminex MagPlex magnetic microspheres for high-throughput spoligotyping of Mycobacterium tuberculosis isolates in Port-au-Prince, Haiti. J. Clin. Microbiol. 2013, 51, 2232–2237. [Google Scholar] [CrossRef]
- Zhang, J.; Abadia, E.; Refregier, G.; Tafaj, S.; Boschiroli, M.L.; Guillard, B.; Andremont, A.; Ruimy, R.; Sola, C. Mycobacterium tuberculosis complex CRISPR genotyping: Improving efficiency, throughput and discriminative power of “spoligotyping” with new spacers and a microbead-based hybridization assay. J. Med. Microbiol. 2010, 59, 285–294. [Google Scholar] [CrossRef]
- Honisch, C.; Mosko, M.; Arnold, C.; Gharbia, S.E.; Diel, R.; Niemann, S. Replacing reverse line blot hybridization spoligotyping of the Mycobacterium tuberculosis complex. J. Clin. Microbiol. 2010, 48, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Jeong, H.J.; Lee, S.M.; Kim, C.M.; Song, E.S.; Park, Y.K.; Bai, G.H.; Lee, E.Y.; Chang, C.L. A DNA chip-based spoligotyping method for the strain identification of Mycobacterium tuberculosis isolates. J. Microbiol. Methods 2007, 68, 430–433. [Google Scholar] [CrossRef]
- Ruettger, A.; Nieter, J.; Skrypnyk, A.; Engelmann, I.; Ziegler, A.; Moser, I.; Monecke, S.; Ehricht, R.; Sachse, K. Rapid spoligotyping of Mycobacterium tuberculosis complex bacteria by use of a microarray system with automatic data processing and assignment. J. Clin. Microbiol. 2012, 50, 2492–2495. [Google Scholar] [CrossRef] [PubMed]
- Bespyatykh, J.A.; Zimenkov, D.V.; Shitikov, E.A.; Kulagina, E.V.; Lapa, S.A.; Gryadunov, D.A.; Ilina, E.N.; Govorun, V.M. Spoligotyping of Mycobacterium tuberculosis complex isolates using hydrogel oligonucleotide microarrays. Infect. Genet. Evol. 2014, 26, 41–46. [Google Scholar] [CrossRef]
- Cabibbe, A.M.; Miotto, P.; Moure, R.; Alcaide, F.; Feuerriegel, S.; Pozzi, G.; Nikolayevskyy, V.; Drobniewski, F.; Niemann, S.; Reither, K.; et al. Lab-on-chip-based platform for fast molecular diagnosis of multidrug-resistant tuberculosis. J. Clin. Microbiol. 2015, 53, 3876–3880. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, H.; Zheng, R.; Kurepina, N.; Kreiswirth, B.N.; Zhao, X.; Xu, Y.; Li, Q.; Diagnostic, M. Spoligotyping of Mycobacterium tuberculosis complex isolates using ligation-based amplification and melting curve analysis. J. Clin. Microbiol. 2016, 54, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.H. The global distribution and phylogeography of Mycobacterium bovis clonal complexes. Infect. Genet. Evol. 2012, 12, 857–865. [Google Scholar] [CrossRef]
- Warren, R.M.; Streicher, E.M.; Sampson, S.L.; van der Spuy, G.D.; Richardson, M.; Nguyen, D.; Behr, M.A.; Victor, T.C.; van Helden, P.D. Microevolution of the Direct Repeat Region of Mycobacterium tuberculosis: Implications for Interpretation of Spoligotyping Data. J. Clin. Microbiol. 2002, 40, 4457–4465. [Google Scholar] [CrossRef]
- Ramazanzadeh, R.; McNerney, R. Variable number of tandem repeats (VNTR) and its application in bacterial epidemiology. Pak. J. Biol. Sci. 2007, 10, 2612–2621. [Google Scholar]
- Frothingham, R.; Meeker-O’Connell, W.A. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology 1998, 144, 1189–1196. [Google Scholar] [CrossRef]
- Frothingham, R. Differentiation of strains in Mycobacterium tuberculosis complex by DNA sequence polymorphisms, including rapid identification of M. bovis BCG. J. Clin. Microbiol. 1995, 33, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Supply, P.; Magdalena, J.; Himpens, S.; Locht, C. Identification of novel intergenic repetitive units in a mycobacterial two-component system operon. Mol. Microbiol. 1997, 26, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Kremer, K.; Van Soolingen, D.; Frothingham, R.; Haas, W.H.; Hermans, P.W.M.; Martín, C.; Palittapongarnpim, P.; Plikaytis, B.B.; Riley, L.W.; Yakrus, M.A.; et al. Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: Interlaboratory study of discriminatory power and reproducibility. J. Clin. Microbiol. 1999, 37, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Barlow, R.E.L.; Gascoyne-binzi, D.M.; Gillespie, S.H.; Dickens, A.; Qamer, S.; Hawkey, P.M. Comparison of variable number tandem repeat and IS6110-restriction fragment length polymorphism analyses for discrimination of high- and low-copy-number IS6110 Mycobacterium tuberculosis isolates. J. Clin. Microbiol. 2001, 39, 2453–2457. [Google Scholar] [CrossRef]
- Filliol, I.; Ferdinand, S.; Negroni, L.; Sola, C.; Rastogi, N. Molecular typing of Mycobacterium tuberculosis based on variable number of tandem DNA repeats used alone and in association with spoligotyping. J. Clin. Microbiol. 2000, 38, 2520–2524. [Google Scholar] [CrossRef]
- Supply, P.; Allix, C.; Lesjean, S.; Cardoso-Oelemann, M.; Rusch-Gerdes, S.; Willery, E.; Savine, E.; de Haas, P.; van Deutekom, H.; Roring, S.; et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 4498–4510. [Google Scholar] [CrossRef]
- Weniger, T.; Krawczyk, J.; Supply, P.; Niemann, S.; Harmsen, D. MIRU-VNTRplus: A web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010, 38, W326–W331. [Google Scholar] [CrossRef][Green Version]
- Hauer, A.; Michelet, L.; De Cruz, K.; Cochard, T.; Branger, M.; Karoui, C.; Henault, S.; Biet, F.; Boschiroli, M.L. MIRU-VNTR allelic variability depends on Mycobacterium bovis clonal group identity. Infect. Genet. Evol. 2016, 45, 165–169. [Google Scholar] [CrossRef]
- Wyllie, D.H.; Davidson, J.A.; Grace Smith, E.; Rathod, P.; Crook, D.W.; Peto, T.E.A.; Robinson, E.; Walker, T.; Campbell, C. A quantitative evaluation of MIRU-VNTR typing against whole-genome sequencing for identifying Mycobacterium tuberculosis transmission: A prospective observational cohort study. EBioMedicine 2018, 34, 122–130. [Google Scholar] [CrossRef]
- Allix-Béguec, C.; Wahl, C.; Hanekom, M.; Nikolayevskyy, V.; Drobniewski, F.; Maeda, S.; Campos-Herrero, I.; Mokrousov, I.; Niemann, S.; Kontsevaya, I.; et al. Proposal of a consensus set of hypervariable mycobacterial interspersed repetitive-unit-variable-number tandem-repeat loci for subtyping of Mycobacterium tuberculosis Beijing isolates. J. Clin. Microbiol. 2014, 52, 164–172. [Google Scholar] [CrossRef]
- Garnier, T.; Eiglmeier, K.; Camus, J.-C.; Medina, N.; Mansoor, H.; Pryor, M.; Duthoy, S.; Grondin, S.; Lacroix, C.; Monsempe, C.; et al. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 2003, 100, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Malone, K.M.; Farrell, D.; Stuber, T.P.; Schubert, O.T. Updated reference genome sequence and annotation of Mycobacterium bovis AF2122/97. Genome Announc. 2017, 5, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Bouso, J.M.; Planet, P.J. Complete nontuberculous mycobacteria whole genomes using an optimized DNA extraction protocol for long-read sequencing. BMC Genom. 2019, 20, 793. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.; Edwards, K.; Desai, M.; Platt, S.; Green, J.; Conway, D. Setup, validation, and quality control of a centralized whole- genome-sequencing laboratory: Lessons learned. J. Clin. Microbiol. 2018, 56, e-00261-18. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, H.; Fei, Z.; Zhan, F.; Gong, X.; Gao, S. Fastq-clean: An optimized pipeline to clean the Illumina sequencing data with quality control. In Proceedings of the 2014 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Belfast, UK, 2–5 November 2014; pp. 44–48. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Hannon, G.J. FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 15 December 2019).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 December 2019).
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, M.T.W.; Wadsworth, M.E.; Staley, L.A.; Hoyt, K.L.; Pickett, B.; Miller, J.; Duce, J.; Kauwe, J.S.K.; Ridge, P.G. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinform. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Pavesi, G. Evaluation of quality assessment protocols for high throughput genome resequencing data. Front. Genet. 2017, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Del Fabbro, C.; Scalabrin, S.; Morgante, M.; Giorgi, F.M. An extensive evaluation of read trimming effects on illumina NGS data analysis. PLoS ONE 2013, 8, e85024. [Google Scholar] [CrossRef]
- Lusk, R.W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data. PLoS ONE 2014, 9, e110808. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Pertea, M.; Zimin, A.V.; Salzberg, S.L. Human contamination in bacterial genomes has created thousands of spurious proteins. Genome Res. 2019, 29, 954–960. [Google Scholar] [CrossRef]
- Merchant, S.; Wood, D.E.; Salzberg, S.L. Unexpected cross-species contamination in genome sequencing projects. Peer J. 2014, 2, e675. [Google Scholar] [CrossRef]
- Goig, G.A.; Garcia-basteiro, A.L.; Cambeve, B. Contaminant DNA in bacterial sequencing experiments is a major source of false genetic variability. BMC Biol. 2020, 18, 748. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Ezewudo, M.; Borens, A.; Chiner-Oms, Á.; Miotto, P.; Chindelevitch, L.; Starks, A.M.; Hanna, D.; Liwski, R.; Zignol, M.; Gilpin, C.; et al. Integrating standardized whole genome sequence analysis with a global Mycobacterium tuberculosis antibiotic resistance knowledgebase. Sci. Rep. 2018, 8, 15382. [Google Scholar] [CrossRef]
- Wyllie, D.H.; Robinson, E.; Peto, T.; Crook, D.W.; Ajileye, A.; Rathod, P.; Allen, R.; Jarrett, L.; Smith, E.G.; Walker, A.S. Identifying mixed Mycobacterium tuberculosis infection and laboratory cross-contamination during mycobacterial sequencing programs. J. Clin. Microbiol. 2018, 56, e00923-18. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pereira, T.T.; Ikuta, C.Y.; Zimpel, C.K.; Camargo, N.C.S.; Filho, A.F.D.S.; Neto, J.S.F.; Heinemann, M.B.; Guimarães, A.M.S. Genome sequencing of Mycobacterium pinnipedii strains: Genetic characterization and evidence of superinfection in a South American sea lion (Otaria flavescens). BMC Genom. 2019, 1030. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Lewin, A.; Metzger, S.; Maetz-Rennsing, K.; Calvignac-Spencer, S.; Nitsche, A.; Dabrowski, P.W.; Radonic, A.; Niemann, S.; Parkhill, J.; et al. Novel Mycobacterium tuberculosis Complex Isolate from a Wild Chimpanzee. Emerg. Infect. Dis. 2013, 19, 969–976. [Google Scholar] [CrossRef]
- Dou, H.; Lin, C.; Ch, Y.; Yang, S.; Chang, J. Lineage-specific SNPs for genotyping of Mycobacterium tuberculosis clinical isolates. Sci. Rep. 2017, 7, 1425. [Google Scholar] [CrossRef]
- Bainomugisa, A.; Lavu, E.; Hiashiri, S.; Majumdar, S.; Honjepari, A.; Moke, R.; Dakulala, P.; Hill-cawthorne, G.A.; Pandey, S.; Marais, B.J.; et al. Multi-clonal evolution of multi-drug-resistant/extensively drug- resistant Mycobacterium tuberculosis in a high-prevalence setting of Papua New Guinea for over three decades. Microb. Genom. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Jajou, R.; Kohl, T.A.; Walker, T.; Norman, A.; Cirillo, D.M.; Tagliani, E.; Niemann, S.; De Neeling, A.; Lillebaek, T.; Anthony, R.M.; et al. Towards standardisation: Comparison of five whole genome sequencing (WGS) analysis pipelines for detection of epidemiologically linked tuberculosis cases. Eurosurveillance 2019, 24. [Google Scholar] [CrossRef]
- Zimpel, C.K.; Brandão, P.E.; de Souza Filho, A.F.; de Souza, R.F.; Ikuta, C.Y.; Ferreira Neto, J.S.; Camargo, N.C.S.; Heinemann, M.B.; Guimarães, A.M.S. Complete Genome Sequencing of Mycobacterium bovis SP38 and Comparative Genomics of Mycobacterium bovis and M. tuberculosis Strains. Front. Microbiol. 2017, 8, 2389. [Google Scholar] [CrossRef]
- Faksri, K.; Xia, E.; Tan, J.H.; Teo, Y.Y.; Ong, R.T.H. In silico region of difference (RD) analysis of Mycobacterium tuberculosis complex from sequence reads using RD-Analyzer. BMC Genom. 2016, 17, 847. [Google Scholar] [CrossRef]
- Hatherell, H.A.; Colijn, C.; Stagg, H.R.; Jackson, C.; Winter, J.R.; Abubakar, I. Interpreting whole genome sequencing for investigating tuberculosis transmission: A systematic review. BMC Med. 2016, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.S.; Behr, M.A. Does choice matter? Reference-based alignment for molecular epidemiology of tuberculosis. J. Clin. Microbiol. 2016, 54, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.S.; Colijn, C.; Cohen, T.; Mathema, B.; Liu, Q.; Bowers, J.; Engelthaler, D.M.; Narechania, A.; Croda, J.; Andrews, J.R. Genomic variant identification methods alter Mycobacterium tuberculosis transmission inference. bioRxiv 2019, 733642. [Google Scholar] [CrossRef]
- Bush, S.J.; Foster, D.; Eyre, D.W.; Clark, E.L.; De Maio, N.; Shaw, L.P.; Stoesser, N.; Peto, T.E.A.; Crook, D.W.; Walker, A.S. Genomic diversity affects the accuracy of bacterial SNP calling pipelines. Gigascience 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Dippenaar, A.; Parsons, S.D.C.; Miller, M.A.; Hlokwe, T.; Gey van Pittius, N.C.; Adroub, S.A.; Abdallah, A.M.; Pain, A.; Warren, R.M.; Michel, A.L.; et al. Progenitor strain introduction of Mycobacterium bovis at the wildlife-livestock interface can lead to clonal expansion of the disease in a single ecosystem. Infect. Genet. Evol. 2017, 51, 235–238. [Google Scholar] [CrossRef]
- Branger, M.; Loux, V.; Cochard, T.; Boschiroli, M.L.; Biet, F.; Michelet, L. The complete genome sequence of Mycobacterium bovis Mb3601, a SB0120 spoligotype strain representative of a new clonal group. Infect. Genet. Evol. 2020, 82, 104309. [Google Scholar] [CrossRef]
- Jandrasits, C.; Kröger, S.; Haas, W.; Renard, B.Y. Computational pan-genome mapping and pairwise SNP-distance improve detection of Mycobacterium tuberculosis transmission clusters. bioRxiv 2019, 15, 1–20. [Google Scholar] [CrossRef]
- Olson, N.D.; Lund, S.P.; Colman, R.E.; Foster, J.T.; Sahl, J.W.; Schupp, J.M.; Keim, P.; Morrow, J.B.; Salit, M.L.; Zook, J.M. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front. Genet. 2015, 6, 235. [Google Scholar] [CrossRef]
- Inouye, M.; Dashnow, H.; Raven, L.A.; Schultz, M.B.; Pope, B.J.; Tomita, T.; Zobel, J.; Holt, K.E. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014, 6, 1–16. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Raczy, C.; Petrovski, R.; Saunders, C.T.; Chorny, I.; Kruglyak, S.; Margulies, E.H.; Chuang, H.Y.; Källberg, M.; Kumar, S.A.; Liao, A.; et al. Isaac: Ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 2013, 29, 2041–2043. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Canzar, S.; Salzberg, S.L. Short read mapping: An algorithmic tour. Proc. IEEE Inst. Electr. Electron. Eng. 2017, 105, 436–458. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv Prepr. 2012, arXiv:1207.3907. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, D.; Kajitani, R.; Gotoh, Y.; Katahira, K.; Okuno, M.; Ogura, Y.; Hayashi, T.; Itoh, T. Evaluation of SNP calling methods for closely related bacterial isolates and a novel high-accuracy pipeline: BactSNP. Microb. Genom. 2019, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pouseele, H.; Supply, P. Accurate whole-genome sequencing-based epidemiological surveillance of Mycobacterium tuberculosis. In Current and Emerging Technologies for the Diagnosis of Microbial Infections; Sails, A., Tang, Y.-W., Eds.; Elsevier Ltd: Oxford, UK, 2015; pp. 359–386. [Google Scholar]
- Vargas, R., Jr.; Freschi, L.; Marin, M.; Epperson, L.E.; Smith, M.; Oussenko, I.; Durbin, D.; Strong, M.; Salfinger, M.; Farhat, M.R. In-host population dynamics of M. tuberculosis during treatment failure. bioRxiv 2019, 6, 726430. [Google Scholar]
- Lee, R.S.; Proulx, J.-F.; McIntosh, F.; Behr, M.A.; Hanage, W.P. Previously undetected super-spreading of Mycobacterium tuberculosis revealed by deep sequencing. Elife 2020, 9, e53245. [Google Scholar] [CrossRef]
- Bryant, J.M.; Harris, S.R.; Parkhill, J.; Dawson, R.; Diacon, A.H.; van Helden, P.; Pym, A.; Mahayiddin, A.A.; Chuchottaworn, C.; Sanne, I.M.; et al. Whole-genome sequencing to establish relapse or re-infection with Mycobacterium tuberculosis: A retrospective observational study. Lancet Respir. Med. 2013, 1, 786–792. [Google Scholar] [CrossRef]
- Sobkowiak, B.; Glynn, J.R.; Houben, R.M.G.J.; Mallard, K.; Phelan, J.E.; Guerra-Assunção, J.A.; Banda, L.; Mzembe, T.; Viveiros, M.; McNerney, R.; et al. Identifying mixed Mycobacterium tuberculosis infections from whole genome sequence data. BMC Genom. 2018, 19, 613. [Google Scholar] [CrossRef]
- Brites, D.; Loiseau, C.; Menardo, F.; Borrell, S.; Boniotti, M.B.; Warren, R.; Dippenaar, A.; Parsons, S.D.C.; Beisel, C.; Behr, M.A.; et al. A new phylogenetic framework for the animal-adapted Mycobacterium tuberculosis complex. Front. Microbiol. 2018, 9, 2820. [Google Scholar] [CrossRef]
- Guerra-Assunção, J.A.; Houben, R.M.G.J.; Crampin, A.C.; Mzembe, T.; Mallard, K.; Coll, F.; Khan, P.; Banda, L.; Chiwaya, A.; Pereira, R.P.A.; et al. Recurrence due to relapse or reinfection with Mycobacterium tuberculosis: A whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up. J. Infect. Dis. 2015, 211, 1154–1163. [Google Scholar] [CrossRef]
- Sandoval-Azuara, S.E.; Muñiz-Salazar, R.; Perea-Jacobo, R.; Robbe-Austerman, S.; Perera-Ortiz, A.; López-Valencia, G.; Bravo, D.M.; Sanchez-Flores, A.; Miranda-Guzmán, D.; Flores-López, C.A.; et al. Whole genome sequencing of Mycobacterium bovis to obtain molecular fingerprints in human and cattle isolates from Baja California, Mexico. Int. J. Infect. Dis. 2017, 63, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Bruning-Fann, C.; Robbe-Austerman, S.; Kaneene, J.; Thomsen, B.; Tilden, J.D., Jr.; Ray, J.; Smith, R.; Fitzgerald, S.; Bolin, S.; O’Brien, D.; et al. Use of whole-genome sequencing and evaluation of the apparent sensitivity and specificity of antemortem tuberculosis tests in the investigation of an unusual outbreak of Mycobacterium bovis infection in a Michigan dairy herd. J. Am. Vet. Med. Assoc. 2017, 251, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Ip, C.L.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef]
- Kato-Maeda, M.; Ho, C.; Passarelli, B.; Banaei, N.; Grinsdale, J.; Flores, L.; Anderson, J.; Murray, M.; Rose, G.; Kawamura, L.M.; et al. Use of whole genome sequencing to determine the microevolution of Mycobacterium tuberculosis during an outbreak. PLoS ONE 2013, 8, e58235. [Google Scholar] [CrossRef]
- Pérez-Lago, L.; Comas, I.; Navarro, Y.; González-Candelas, F.; Herranz, M.; Bouza, E.; García-De-Viedma, D. Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: Potential impact on the inference of tuberculosis transmission. J. Infect. Dis. 2014, 209, 98–108. [Google Scholar] [CrossRef]
- Luo, T.; Yang, C.; Peng, Y.; Lu, L.; Sun, G.; Wu, J.; Jin, X.; Hong, J.; Li, F.; Mei, J.; et al. Whole-genome sequencing to detect recent transmission of Mycobacterium tuberculosis in settings with a high burden of tuberculosis. Tuberculosis 2014, 94, 434–440. [Google Scholar] [CrossRef]
- Comas, I.; Hailu, E.; Kiros, T.; Bekele, S.; Mekonnen, W.; Gumi, B.; Tschopp, R.; Ameni, G.; Hewinson, R.G.; Robertson, B.D.; et al. Population Genomics of Mycobacterium tuberculosis in Ethiopia Contradicts the Virgin Soil Hypothesis for Human Tuberculosis in Sub-Saharan Africa. Curr. Biol. 2015, 25, 3260–3266. [Google Scholar] [CrossRef]
- Faksri, K.; Xia, E.; Ong, R.T.-H.; Tan, J.H.; Nonghanphithak, D.; Makhao, N.; Thamnongdee, N.; Thanormchat, A.; Phurattanakornkul, A.; Rattanarangsee, S.; et al. Comparative whole-genome sequence analysis of Mycobacterium tuberculosis isolated from tuberculous meningitis and pulmonary tuberculosis patients. Sci. Rep. 2018, 8, 4910. [Google Scholar] [CrossRef]
- Trewby, H.; Wright, D.; Breadon, E.L.; Lycett, S.J.; Mallon, T.R.; McCormick, C.; Johnson, P.; Orton, R.J.; Allen, A.R.; Galbraith, J.; et al. Use of bacterial whole-genome sequencing to investigate local persistence and spread in bovine tuberculosis. Epidemics 2016, 14, 26–35. [Google Scholar] [CrossRef]
- Cohen, T.; van Helden, P.D.; Wilson, D.; Colijn, C.; McLaughlin, M.M.; Abubakar, I.; Warren, R.M. Mixed-strain Mycobacterium tuberculosis infections and the implications for tuberculosis treatment and control. Clin. Microbiol. Rev. 2012, 25, 708–719. [Google Scholar] [CrossRef]
- Egbe, N.F.; Muwonge, A.; Ndip, L.; Kelly, R.F.; Sander, M.; Tanya, V.; Ngwa, V.N.; Handel, I.G.; Novak, A.; Ngandalo, R.; et al. Molecular epidemiology of Mycobacterium bovis in Cameroon. Sci. Rep. 2017, 7, 4652. [Google Scholar] [CrossRef] [PubMed]
- Ghielmetti, G.; Coscolla, M.; Ruetten, M.; Friedel, U.; Loiseau, C.; Feldmann, J.; Steinmetz, H.W.; Stucki, D.; Gagneux, S. Tuberculosis in Swiss captive Asian elephants: Microevolution of Mycobacterium tuberculosis characterized by multilocus variable-number tandem-repeat analysis and whole-genome sequencing. Sci. Rep. 2017, 7, 14647. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Via, L.E.; Luo, T.; Liang, L.; Liu, X.; Wu, S.; Shen, Q.; Wei, W.; Ruan, X.; Yuan, X.; et al. Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci. Rep. 2015, 5, 17507. [Google Scholar] [CrossRef] [PubMed]
- Ssengooba, W.; de Jong, B.C.; Joloba, M.L.; Cobelens, F.G.; Meehan, C.J. Whole genome sequencing reveals mycobacterial microevolution among concurrent isolates from sputum and blood in HIV infected TB patients. BMC Infect. Dis. 2016, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, T.D.; Wilson, D.; Misra, R.; Xiong, L.L.; Moodley, P.; Cohen, T.; Kishony, R.; Author, N.M. Genomic diversity in autopsy samples reveals within-host dissemination of HIV-associated M. tuberculosis HHS Public Access Author manuscript. Nat. Med. 2016, 22, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S.; et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; O’Hare, A.; Wright, D.; Mallon, T.; McCormick, C.; Orton, R.J.; McDowell, S.; Trewby, H.; Skuce, R.A.; Kao, R.R. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 2012, 8, e1003008. [Google Scholar] [CrossRef]
- Guerra-Assunção, J.A.; Crampin, A.C.; Houben, R.M.G.J.; Mzembe, T.; Mallard, K.; Coll, F.; Khan, P.; Banda, L.; Chiwaya, A.; Pereira, R.P.A.; et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife 2015, 2015, e05166. [Google Scholar]
- Michelet, L.; Conde, C.; Branger, M.; Cochard, T.; Biet, F.; Boschiroli, M.L. Transmission Network of Deer-Borne Mycobacterium bovis Infection Revealed by a WGS Approach. Microorganisms 2019, 687. [Google Scholar] [CrossRef]
- Clark, T.G.; Mallard, K.; Coll, F.; Preston, M.; Assefa, S.; Harris, D.; Ogwang, S.; Mumbowa, F.; Kirenga, B.; O’Sullivan, D.M.; et al. Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing. PLoS ONE 2013, 8, e83012. [Google Scholar] [CrossRef]
- Lee, R.S.; Radomski, N.; Proulx, J.F.; Manry, J.; McIntosh, F.; Desjardins, F.; Soualhine, H.; Domenech, P.; Reed, M.B.; Menzies, D.; et al. Reemergence and amplification of tuberculosis in the Canadian Arctic. J. Infect. Dis. 2015, 211, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.M.; Lalor, M.K.; Broda, A.; Ortega, L.S.; Parker, L.; Churchill, S.; Bennett, K.; Golubchik, T.; Giess, A.P.; Del, C.; et al. Assessment of Mycobacterium tuberculosis transmission in Oxfordshire, UK, 2007—2012, with whole pathogen genome sequences: An observational study. Lancet Infect. Dis. 2015, 2, 285–292. [Google Scholar]
- Witney, A.A.; Gould, K.A.; Arnold, A.; Coleman, D.; Delgado, R.; Dhillon, J.; Pond, M.J.; Pope, C.F.; Planche, T.D.; Stoker, N.G.; et al. Clinical application of whole-genome sequencing to inform treatment for multidrug-resistant tuberculosis cases. J. Clin. Microbiol. 2015, 53, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Acosta, F.; Chernyaeva, E.; Mendoza, L.; Sambrano, D.; Correa, R.; Rotkevich, M.; Tarté, M.; Hernández, H.; Velazco, B.; de Escobar, C.; et al. Mycobacterium bovis in Panama, 2013. Emerg. Infect. Dis. 2015, 21, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Outhred, A.C.; Holmes, N.; Sadsad, R.; Martinez, E.; Jelfs, P.; Hill-Cawthorne, G.A.; Gilbert, G.L.; Marais, B.J.; Sintchenko, V. Identifying likely transmission pathways within a 10-year community outbreak of tuberculosis by high-depth whole genome sequencing. PLoS ONE 2016, 11, e0150550. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.A.; Diel, R.; Harmsen, D.; Rothgänger, J.; Meywald Walter, K.; Merker, M.; Weniger, T.; Niemann, S. Whole-genome-based Mycobacterium tuberculosis surveillance: A standardized, portable, and expandable approach. J. Clin. Microbiol. 2014, 52, 2479–2486. [Google Scholar] [CrossRef]
- Kohl, T.A.; Harmsen, D.; Rothgänger, J.; Walker, T.; Diel, R.; Niemann, S. Harmonized genome wide typing of tubercle bacilli using a web-based gene-by-gene nomenclature system. EBioMedicine 2018, 34, 131–138. [Google Scholar] [CrossRef]
- Ridom SeqSphere+ Software. Available online: https://www.ridom.de/seqsphere/ (accessed on 8 January 2020).
- Bionumerics. Bionumerics for Whole Genome Multi Locus Sequence Typing. Available online: http://www.applied-maths.com/applications/wgmlst (accessed on 8 January 2020).
- Kavvas, E.S.; Catoiu, E.; Mih, N.; Yurkovich, J.T.; Seif, Y.; Dillon, N.; Heckmann, D.; Anand, A.; Yang, L.; Nizet, V.; et al. Machine learning and structural analysis of Mycobacterium tuberculosis pan-genome identifies genetic signatures of antibiotic resistance. Nat. Commun. 2018, 9, 4306. [Google Scholar] [CrossRef]
- Periwal, V.; Patowary, A.; Vellarikkal, S.K.; Gupta, A.; Singh, M.; Mittal, A.; Jeyapaul, S.; Chauhan, R.K.; Singh, A.V.; Singh, P.K.; et al. Comparative Whole-Genome Analysis of Clinical Isolates Reveals Characteristic Architecture of Mycobacterium tuberculosis Pangenome. PLoS ONE 2015, 10, e0122979. [Google Scholar] [CrossRef]
- Anzai, E.K. Sequenciamento do Genoma Completo do Mycobacterium bovis Como Instrumento de Sistema de Vigilância no Estado de Santa Catarina; University of São Paulo: Sao Paulo, Brazil, 2019. [Google Scholar]
- Lasserre, M.; Fresia, P.; Greif, G.; Iraola, G.; Castro-Ramos, M.; Juambeltz, A.; Nuñez, Á.; Naya, H.; Robello, C.; Berná, L. Whole genome sequencing of the monomorphic pathogen Mycobacterium bovis reveals local differentiation of cattle clinical isolates. BMC Genom. 2018, 19, 2. [Google Scholar] [CrossRef]
- Otchere, I.D.; van Tonder, A.J.; Asante-Poku, A.; Sánchez-Busó, L.; Coscollá, M.; Osei-Wusu, S.; Asare, P.; Aboagye, S.Y.; Ekuban, S.A.; Yahayah, A.I.; et al. Molecular epidemiology and whole genome sequencing analysis of clinical Mycobacterium bovis from Ghana. PLoS ONE 2019, 14, e0209395. [Google Scholar] [CrossRef]
- Hauer, A.; Michelet, L.; Cochard, T.; Branger, M.; Nunez, J.; Boschiroli, M.-L.; Biet, F. Accurate phylogenetic relationships among Mycobacterium bovis strains circulating in France based on whole genome sequencing and single nucleotide polymorphism analysis. Front. Microbiol. 2019, 10, 955. [Google Scholar] [CrossRef] [PubMed]
- Patané, J.S.L.; Martins, J.; Castelão, A.B.; Nishibe, C.; Montera, L.; Bigi, F.; Zumárraga, M.J.; Cataldi, A.A.; Junior, A.F.; Roxo, E.; et al. Patterns and processes of Mycobacterium bovis evolution revealed by phylogenomic analyses. Genome Biol. Evol. 2017, 9, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Ypma, R.J.F.; van Ballegooijen, W.M.; Wallinga, J. Relating phylogenetic trees to transmission trees of infectious disease outbreaks. Genetics 2013, 195, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Eggo, R.M.; Dodd, P.J.; Balloux, F. Reconstructing disease outbreaks from genetic data: A graph approach. Heredity 2011, 106, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Zojer, M.; Schuster, L.N.; Schulz, F.; Pfundner, A.; Horn, M.; Rattei, T. Variant profiling of evolving prokaryotic populations. Peer J. 2017, 5, e2997. [Google Scholar] [CrossRef] [PubMed]
- Persi, E.; Wolf, Y.I.; Koonin, E.V. Positive and strongly relaxed purifying selection drive the evolution of repeats in proteins. Nat. Commun. 2016, 7, 13570. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Laehnemann, D.; Borkhardt, A.; McHardy, A.C. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief. Bioinform. 2016, 17, 154–179. [Google Scholar] [CrossRef]
- Fu, S.; Wang, A.; Au, K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Coll, F.; Mallard, K.; Preston, M.D.; Bentley, S.; Parkhill, J.; McNerney, R.; Martin, N.; Clark, T.G. SpolPred: Rapid and accurate prediction of Mycobacterium tuberculosis spoligotypes from short genomic sequences. Bioinformatics 2012, 28, 2991–2993. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Teo, Y.-Y.; Ong, R.T.-H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Guyeux, C.; Sola, C.; Refrégier, G. Exhaustive reconstruction of the CRISPR locus in Mycobacterium tuberculosis complex using short reads. bioRxiv 2019. [Google Scholar] [CrossRef]
- Rajwani, R.; Shehzad, S.; Siu, G.K.H. MIRU-profiler: A rapid tool for determination of 24-loci MIRU-VNTR profiles from assembled genomes of Mycobacterium tuberculosis. Peer J. 2018, 6, e5090. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Y.; Ong, R.T.H. MIRUReader: MIRU-VNTR typing directly from long sequencing reads. Bioinformatics 2020, 36, 1625–1626. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.B.; Lin, P.L.; Chase, M.R.; Shah, R.R.; Iartchouk, O.; Galagan, J.; Mohaideen, N.; Ioerger, T.R.; Sacchettini, J.C.; Lipsitch, M.; et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 2011, 43, 482–486. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO). Challanges of Animal Health Information Systems And Surveillance for Animal Diseases And Zoonoses; FAO Animal Profuction and Health Proceedings; FAO: Rome, Italy, 2011; pp. 1–124. [Google Scholar]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef]
- Timme, R.E.; Rand, H.; Leon, M.S.; Hoffmann, M.; Strain, E.; Allard, M.; Roberson, D.; Baugher, J.D. GenomeTrakr proficiency testing for foodborne pathogen surveillance: An exercise from 2015. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Comin, A.; Grewar, J.; van Schaik, G.; Schwermer, H.; Paré, J.; El Allaki, F.; Drewe, J.; Lopes Antunes, A.C.; Estberg, L.; Horan, M.; et al. Development of reporting guidelines for animal health surveillance—AHSURED. Front. Vet. Sci. 2019, 6, 426. [Google Scholar] [CrossRef]
- Gardy, J.L.; Johnston, J.C.; Sui, S.J.H.; Cook, V.J.; Shah, L.; Brodkin, E.; Rempel, S.; Moore, R.; Zhao, Y.; Holt, R.; et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 2011, 364, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Jajou, R.; de Neeling, A.; van Hunen, R.; de Vries, G.; Schimmel, H.; Mulder, A.; Anthony, R.; van der Hoek, W.; van Soolingen, D. Epidemiological links between tuberculosis cases identified twice as efficiently by whole genome sequencing than conventional molecular typing. PLoS ONE 2018, 13, e0195413. [Google Scholar] [CrossRef] [PubMed]
- Price-Carter, M.; Rooker, S.; Collins, D.M. Comparison of 45 variable number tandem repeat (VNTR) and two direct repeat (DR) assays to restriction endonuclease analysis for typing isolates of Mycobacterium bovis. Vet. Microbiol. 2011, 150, 107–114. [Google Scholar] [CrossRef]
- Collins, D.M. DNA typing of Mycobacterium bovis strains from the castlepoint area of the Wairarapa. N. Z. Vet. J. 1999, 47, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Cao, R.; Tian, M.; Zhang, X.; Zhang, X.; Li, Y.; Xu, Y.; Fan, W.; Huang, B.; Li, C. Evaluation of Spoligotyping and MIRU-VNTR for Mycobacterium bovis in Xinjiang, China. Res. Vet. Sci. 2012, 92, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Campos, S.; Aranaz, A.; De Juan, L.; Sáez-Llorente, J.L.; Romero, B.; Bezos, J.; Jiménez, A.; Mateos, A.; Domínguez, L. Limitations of spoligotyping and variable-number tandem-repeat typing for molecular tracing of Mycobacterium bovis in a high-diversity setting. J. Clin. Microbiol. 2011, 49, 3361–3364. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef]
- Casali, N.; Broda, A.; Harris, S.R.; Parkhill, J.; Brown, T.; Drobniewski, F. Whole genome sequence analysis of a large isoniazid-resistant tuberculosis outbreak in London: A retrospective observational study. PLoS Med. 2016, 13, e1002137. [Google Scholar] [CrossRef]
- Bryant, J.M.; Schürch, A.C.; van Deutekom, H.; Harris, S.R.; de Beer, J.L.; de Jager, V.; Kremer, K.; van Hijum, S.A.F.T.; Siezen, R.J.; Borgdorff, M.; et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect. Dis. 2013, 13, 110. [Google Scholar] [CrossRef]
- Nikolayevskyy, V.; Kranzer, K.; Niemann, S.; Drobniewski, F. Whole genome sequencing of Mycobacterium tuberculosis for detection of recent transmission and tracing outbreaks: A systematic review. Tuberculosis 2016, 98, 77–85. [Google Scholar] [CrossRef]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Brites, D.; Gagneux, S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 2015, 264, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Garcia-Pelayo, M.C.; Müller, B.; Hailu, E.; Asiimwe, B.; Kremer, K.; Dale, J.; Boniotti, M.B.; Rodriguez, S.; Hilty, M.; et al. African 2, a clonal complex of Mycobacterium bovis epidemiologically important in East Africa. J. Bacteriol. 2011, 193, 670–678. [Google Scholar] [CrossRef]
- Müller, B.; Hilty, M.; Berg, S.; Garcia-Pelayo, M.C.; Dale, J.; Boschiroli, M.L.; Cadmus, S.; Ngandolo, B.N.R.; Godreuil, S.; Diguimbaye-Djaibé, C.; et al. African 1, an epidemiologically important clonal complex of Mycobacterium bovis dominant in Mali, Nigeria, Cameroon, and Chad. J. Bacteriol. 2009, 191, 1951–1960. [Google Scholar] [CrossRef]
- Smith, N.H.; Berg, S.; Dale, J.; Allen, A.; Rodriguez, S.; Romero, B.; Matos, F.; Ghebremichael, S.; Karoui, C.; Donati, C.; et al. European 1: A globally important clonal complex of Mycobacterium bovis. Infect. Genet. Evol. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Campos, S.; Schürch, A.C.; Dale, J.; Lohan, A.J.; Cunha, M.V.; Botelho, A.; De Cruz, K.; Boschiroli, M.L.; Boniotti, M.B.; Pacciarini, M.; et al. European 2—A clonal complex of Mycobacterium bovis dominant in the Iberian Peninsula. Infect. Genet. Evol. 2012, 12, 866–872. [Google Scholar] [CrossRef]
- Rodríguez, S.; Bezos, J.; Romero, B.; de Juan, L.; Álvarez, J.; Castellanos, E.; Moya, N.; Lozano, F.; Tariq Javed, M.; Sáez-Llorente, J.L.; et al. Mycobacterium caprae infection in livestock and wildlife, Spain. Emerg. Infect. Dis. 2011, 17, 532–535. [Google Scholar] [CrossRef]
- Pate, M.; Švara, T.; Gombač, M.; Paller, T.; Žolnir-Dovč, M.; Emeršič, I.; Prodinger, W.M.; Bartoš, M.; Zdovc, I.; Krt, B.; et al. Outbreak of tuberculosis caused by Mycobacterium caprae in a zoological garden. J. Vet. Med. Ser. B Infect. Dis. Vet. Public Heal. 2006, 53, 387–392. [Google Scholar] [CrossRef]
- Krajewska, M.; Zabost, A.; Welz, M.; Lipiec, M.; Orłowska, B.; Anusz, K.; Brewczyński, P.; Augustynowicz-Kopeć, E.; Szulowski, K.; Bielecki, W.; et al. Transmission of Mycobacterium caprae in a herd of European bison in the Bieszczady Mountains, Southern Poland. Eur. J. Wildl. Res. 2015, 61, 429–433. [Google Scholar] [CrossRef]
- Sahraoui, N.; Müller, B.; Guetarni, D.; Boulahbal, F.; Yala, D.; Ouzrout, R.; Berg, S.; Smith, N.H.; Zinsstag, J. Molecular characterization of Mycobacterium bovis strains isolated from cattle slaughtered at two abattoirs in Algeria. BMC Vet. Res. 2009, 5, 4. [Google Scholar] [CrossRef]
- Yahyaoui-Azami, H.; Aboukhassib, H.; Bouslikhane, M.; Berrada, J.; Rami, S.; Reinhard, M.; Gagneux, S.; Feldmann, J.; Borrell, S.; Zinsstag, J. Molecular characterization of bovine tuberculosis strains in two slaughterhouses in Morocco. BMC Vet. Res. 2017, 13, 272. [Google Scholar] [CrossRef]
- Yoshida, S.; Suga, S.; Ishikawa, S.; Mukai, Y.; Tsuyuguchi, K.; Inoue, Y.; Yamamoto, T.; Wada, T.; Study, T. Mycobacterium caprae infection in a captive Borneo elephant, Japan. Emerg. Infect. Dis. 2018, 24, 1937–1940. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.C.; Srinivasan, S.; Schilling, M.A.; Stubre, T.; Danchuk, S.N.; Michael, J.S.; Venkatesan, M.; Bansal, N.; Mann, S.; Jindal, N.; et al. Zoonotic tuberculosis in India: Looking beyond Mycobacterium bovis. bioRxiv 2019. [Google Scholar] [CrossRef]
- van Ingen, J.; Rahim, Z.; Mulder, A.; Boeree, M.J.; Simeone, R.; Brosch, R.; van Soolingen, D. Characterization of Mycobacterium orygis as M. tuberculosis complex subspecies. Emerg. Infect. Dis. 2012, 18, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Rahim, Z.; Thapa, J.; Fukushima, Y.; van der Zanden, A.G.M.; Gordon, S.V.; Suzuki, Y.; Nakajima, C. Tuberculosis caused by Mycobacterium orygis in dairy cattle and captured monkeys in Bangladesh: A new scenario of tuberculosis in South Asia. Transbound. Emerg. Dis. 2017, 64, 1965–1969. [Google Scholar] [CrossRef] [PubMed]
- O’Halloran, C.; Hope, J.C.; Dobromylskyj, M.; Burr, P.; McDonald, K.; Rhodes, S.; Roberts, T.; Dampney, R.; De la Rua-Domenech, R.; Robinson, N.; et al. An outbreak of tuberculosis due to Mycobacterium bovis infection in a pack of English Foxhounds (2016–2017). Transbound. Emerg. Dis. 2018, 65, 1872–1884. [Google Scholar] [CrossRef]
- McGill, I.; Saunders, R.; Eastwood, B.; Menache, A.; Dalzell, F.; Hill, S.; Irving, B.; Knight, A.; Jones, M. Mycobacterium bovis tuberculosis in hunting hounds. Vet. Rec. 2018, 183, 387–388. [Google Scholar] [CrossRef]
- Miller, M.A.; Buss, P.; Roos, E.O.; Hausler, G.; Dippenaar, A.; Mitchell, E.; van Schalkwyk, L.; Robbe-Austerman, S.; Waters, W.R.; Sikar-Gang, A.; et al. Fatal tuberculosis in a free-ranging African elephant and one health Implications of human pathogens in wildlife. Front. Vet. Sci. 2019, 6, 18. [Google Scholar] [CrossRef]
- Zachariah, A.; Pandiyan, J.; Madhavilatha, G.K.; Mundayoor, S.; Chandramohan, B.; Sajesh, P.K.; Santhosh, S.; Mikota, S.K. Mycobacterium tuberculosis in wild Asian elephants, southern India. Emerg. Infect. Dis. 2017, 23, 504–506. [Google Scholar] [CrossRef]
- Cui, H.H.; Erkkila, T.; Chain, P.S.G.; Vuyisich, M. Building international genomics collaboration for global health security. Front. Public Heal. 2015, 3, 264. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pipeline Step | Areas in Need of Further Exploration |
|---|---|
| Bacterial isolation and sequencing | Methodologies to assess the possibility of cross-contamination with MTBC isolates |
| Quality assessment of entry data (FASTQ) | Comparison of protocols with different parameters or stringency levels of read trimming and filtering, reference mapping, removal of PCR duplicates, minimum acceptable median read length, contaminants handling, etc. * |
| Read processing | Choice of reference genome Parameters of read mapping (e.g., realignment around indels) Parameters of variant calling How to handle low quality variant calls How to detect and handle variants within repetitive areas Methodologies for detection of mixed-sample (number of reads supporting an allele and number of acceptable heterozygous sites based on established parameters of variant calling) |
| Transmission cluster detection | Comparison and/or development of different approaches: SNP-count, cgMLST, pgMLST, phylogenetic inferences |
| Data reporting | Standardization of WGS data reporting to end-users |
| Validation and inter-laboratory quality control | Validation datasets (of bacterial isolates and genomes) Protocols for inter-laboratory standardization (from bacterial isolation to sequencing) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimaraes, A.M.S.; Zimpel, C.K. Mycobacterium bovis: From Genotyping to Genome Sequencing. Microorganisms 2020, 8, 667. https://doi.org/10.3390/microorganisms8050667
Guimaraes AMS, Zimpel CK. Mycobacterium bovis: From Genotyping to Genome Sequencing. Microorganisms. 2020; 8(5):667. https://doi.org/10.3390/microorganisms8050667
Chicago/Turabian StyleGuimaraes, Ana M. S., and Cristina K. Zimpel. 2020. "Mycobacterium bovis: From Genotyping to Genome Sequencing" Microorganisms 8, no. 5: 667. https://doi.org/10.3390/microorganisms8050667
APA StyleGuimaraes, A. M. S., & Zimpel, C. K. (2020). Mycobacterium bovis: From Genotyping to Genome Sequencing. Microorganisms, 8(5), 667. https://doi.org/10.3390/microorganisms8050667