The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design


Abstract
1. Introduction
2. Herpesviruses
3. Herpesviridae and Human Health Considerations
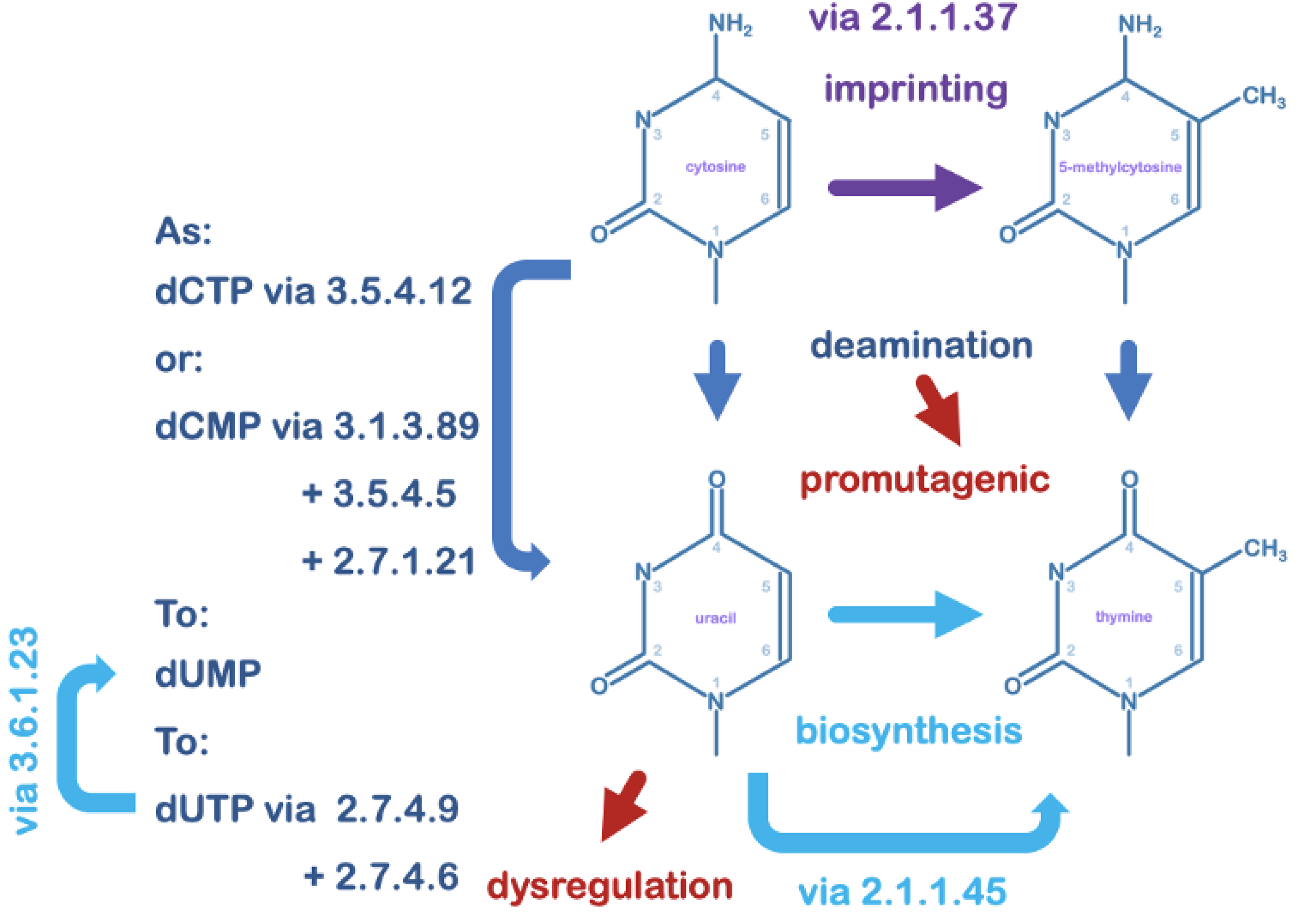
4. Uracil in DNA and its Relevance to Viruses
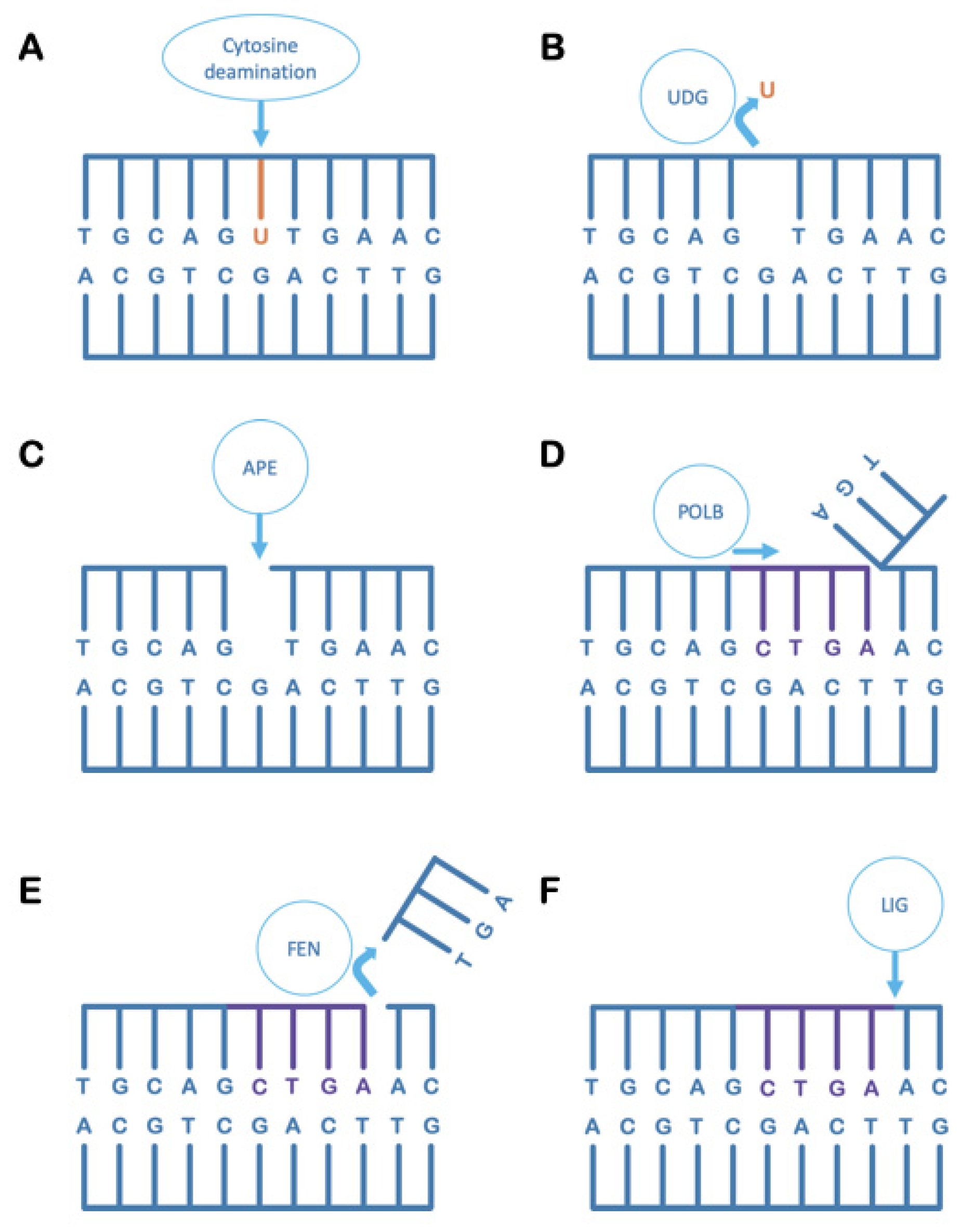
5. Uracil-DNA Glycosylase in the Maintenance of DNA Integrity
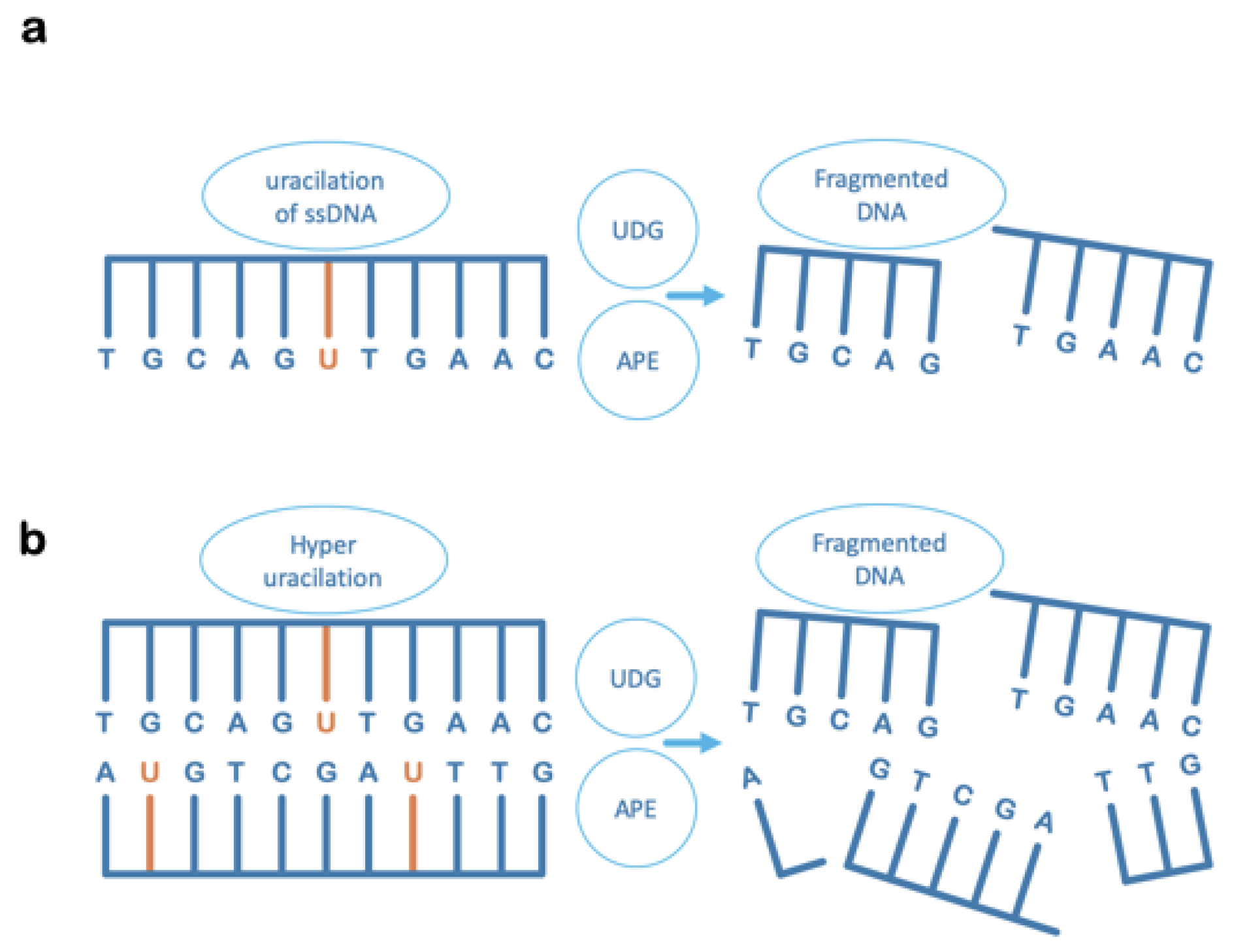
6. Uracil-DNA Glycosylase Can Also Contribute to Irreversible DNA Damage
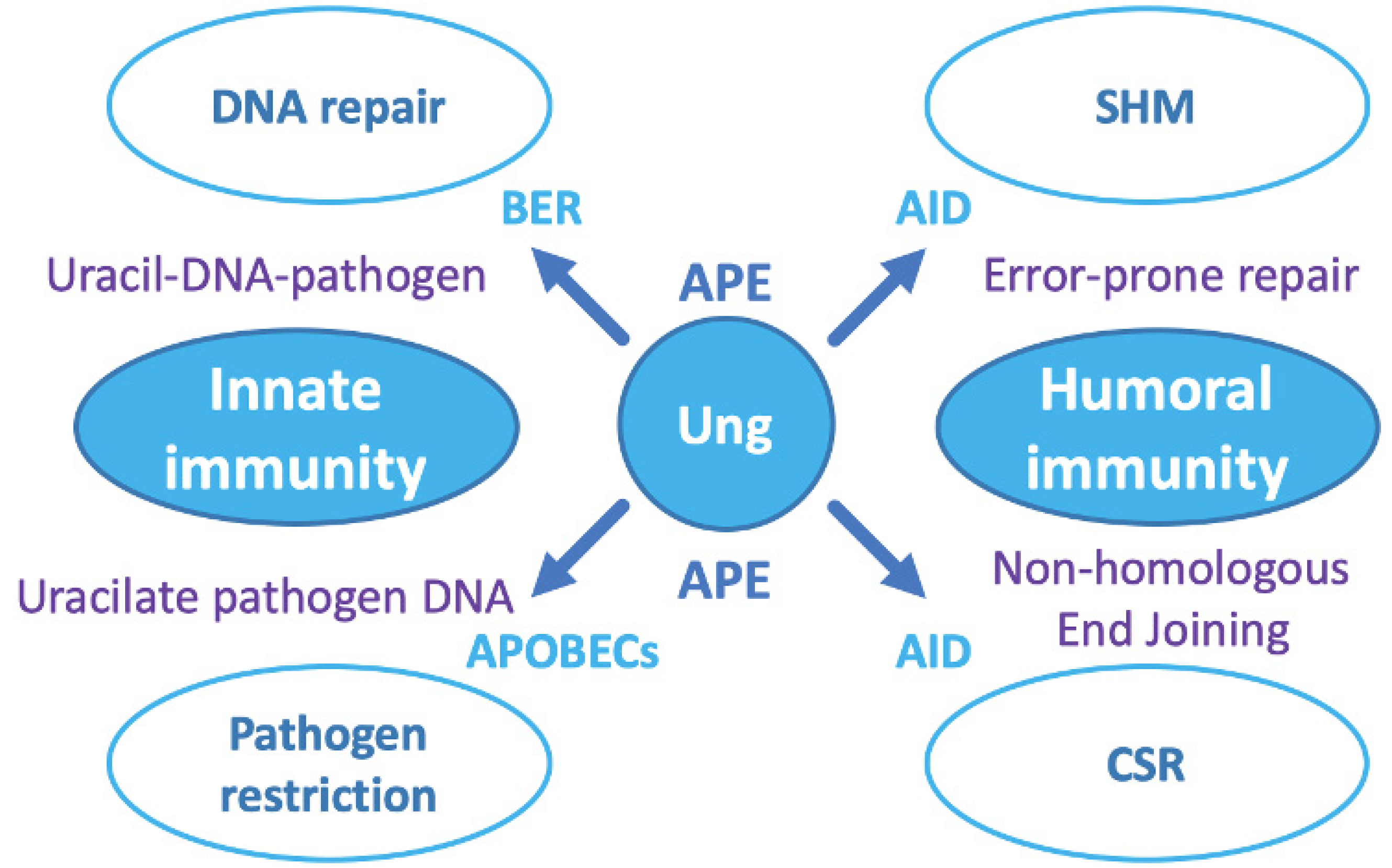
7. The Ung-Type Uracil-DNA Glycosylase Is Central to the Host Pathogen Response
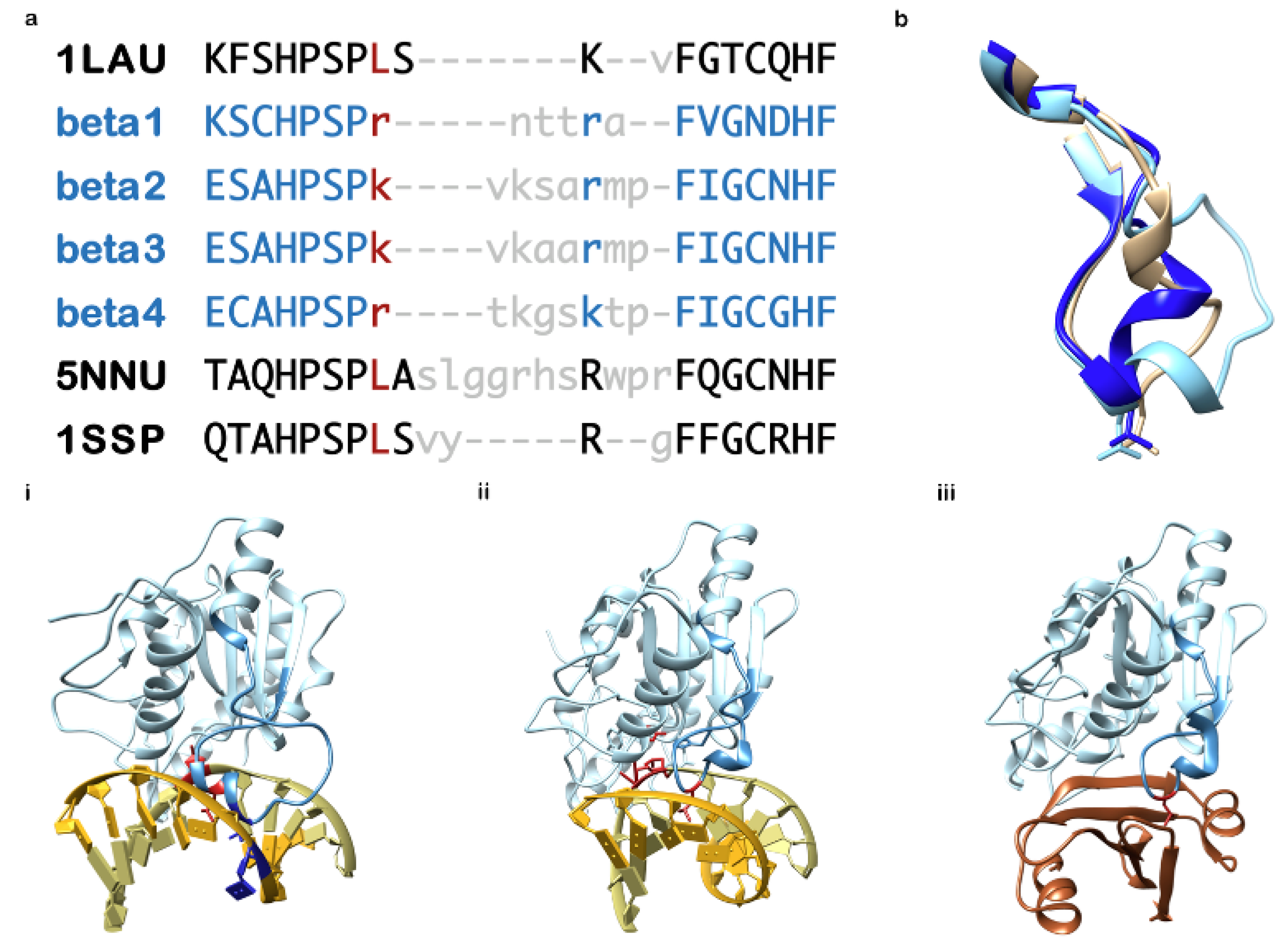
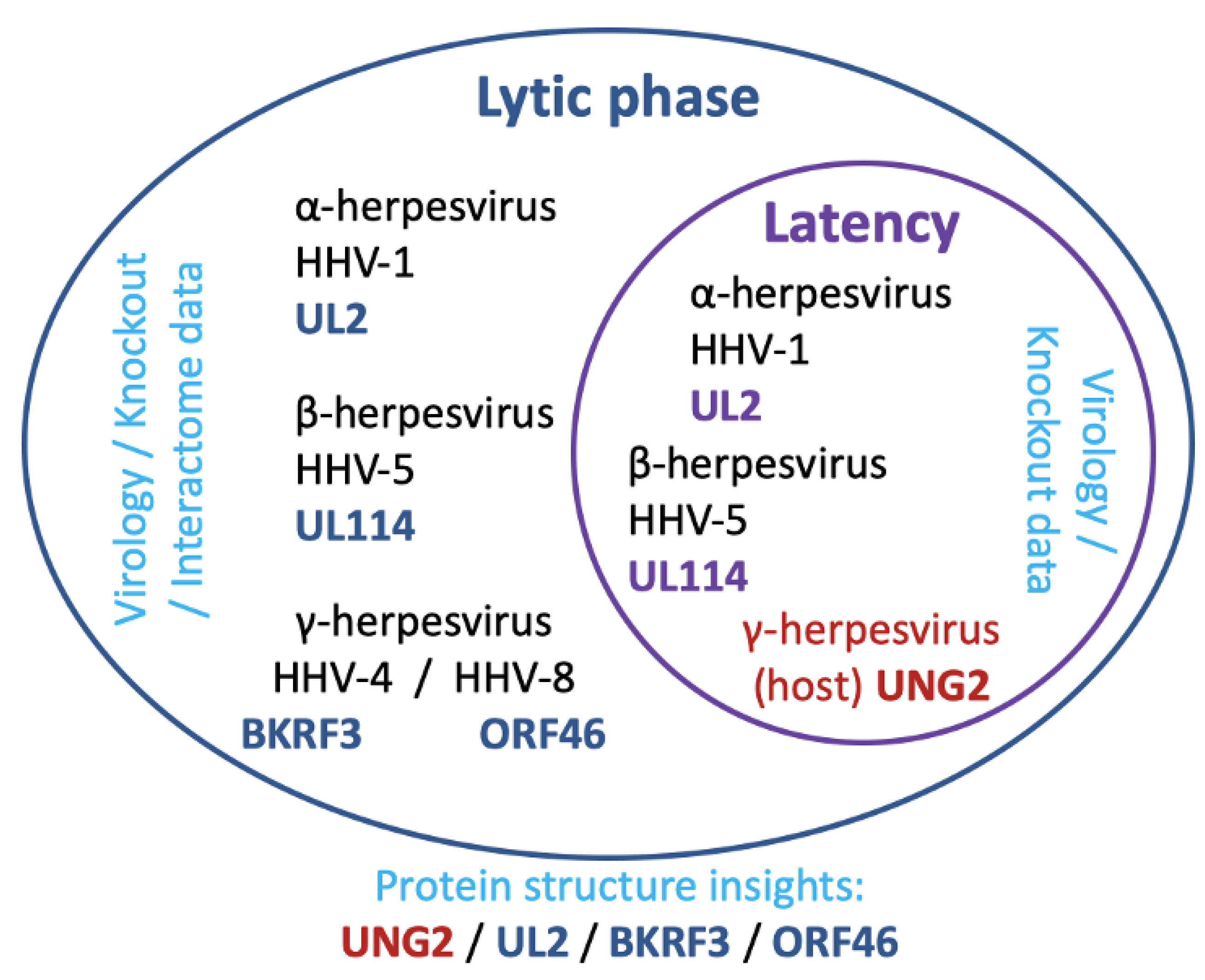
8. Herpesviruses and Ung, a Surprising Relationship and Remarkable Exaptation
9. Uracil in DNA and Its Potential Significance for Herpesviruses in Latency
10. Tropism of Herpesviruses as a Factor in Consideration of Targeting Ung
11. The Reported Roles of Ung in Herpesvirus Fitness
12. A consideration of Specific Selectivity of Novel Chemical Entities for Herpesvirus Ung
13. Summary and Conclusions
Funding
Conflicts of Interest
References
- Arvin, A.; Campadelli-Fiume, G.; Mocarski, E.; Moore, P.S.; Roizman, B.; Whitley, R.; Yamanishi, K. Human Herpesviruses—Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; ISBN-13: 978-0-521-82714-0. [Google Scholar]
- Field, H.J.; Vere Hodge, R.A. Recent developments in anti-herpesvirus drugs. Br. Med. Bull. 2013, 106, 213–249. [Google Scholar] [CrossRef] [PubMed]
- Beswick, T.S.L. The Origin and the Use of the Word Herpes. Med. Hist. 1962, 6, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, B.B.; Flamand, L. Chromosomally integrated HHV-6: Impact on virus, cell and organismal biology. Curr. Opin. Virol. 2014, 9, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Human herpesviruses 6A, 6B, and 7. Microbiol. Spectrum. 2016, 4. [Google Scholar] [CrossRef]
- Berger, J.R.; Houff, S. Neurological Complications of Herpes Simplex Virus Type 2 Infection. Arch. Neurol. 2008, 65, 596–600. [Google Scholar] [CrossRef]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell Infect. Microbiol. 2018, 25, 177. [Google Scholar] [CrossRef]
- Marty, F.M.; Ljungman, P.; Papanicolaou, G.A.; Winston, D.J.; Chemaly, R.F.; Strasfeld, L.; Jo-Anne, H.Y.; Rodriguez, T.; Maertens, J.; Schmitt, M.; et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: A phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2011, 11, 284–292. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Shapiro, R.; Klein, R.S. The deamination of cytidine and cytosine by acidic buffer solutions. Mutagenic implications. Biochemistry 1966, 5, 2358–2362. [Google Scholar] [CrossRef]
- Lindahl, T.; Nyberg, B. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry 1974, 13, 3405–3410. [Google Scholar] [CrossRef]
- Chen, H.; Shaw, B.R. Kinetics of bisulfite-induced cytosine deamination in single-stranded DNA. Biochemistry 1993, 32, 3535–3539. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Zhang, X.Y.; Inamdar, N.M. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat. Res. 1990, 238, 277–286. [Google Scholar] [CrossRef]
- Shen, J.C.; Rideout III, W.M.; Jones, P.A. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994, 22, 972–976. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-e17. [Google Scholar] [CrossRef] [PubMed]
- Chon, J.; Field, M.S.; Stover, P.J. Deoxyuracil in DNA and disease: Genomic signal or managed situation? DNA Repair (Amst.) 2019, 77, 36–44. [Google Scholar] [CrossRef]
- Vértessy, B.G.; Tóth, J. Keeping Uracil Out of DNA: Physiological Role, Structure and Catalytic Mechanism of dUTPases. Acc. Chem. Res. 2009, 42, 97–106. [Google Scholar] [CrossRef]
- Hafez, A.; Messinger, J.; McFadden, K.; Fenyofalvi, G.; Shepard, C.N.; Lenzi, G.M.; Kim, B.; Luftig, M.A. Limited nucleotide pools restrict Epstein–Barr virus-mediated B-cell immortalization. Oncogenesis 2017, 6, e349. [Google Scholar] [CrossRef]
- Julias, J.G.; Pathak, V.K. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate. J. Virol. 1998, 72, 7941–7949. [Google Scholar] [CrossRef]
- Caposio, P.; Riera, L.; Hahn, G.; Landolfo, S.; Gribaudo, G. Evidence that the human cytomegalovirus 46-kDa UL72 protein is not an active dUTPase but a late protein dispensable for replication in fibroblasts. Virology 2004, 325, 264–276. [Google Scholar] [CrossRef]
- Ivarie, R. Thymine methyls and DNA-protein interactions. Nucleic Acids Res. 1987, 15, 9975–9983. [Google Scholar] [CrossRef] [PubMed]
- Verri, A.P.; Mazzarello, G.; Biamonti, S.; Spadari, S.; Focher, F. The specific binding of nuclear protein(s) to the cAMP responsive element (CRE) sequence (TGACGTCA) is reduced by the misincorporation of U and increased by the deamination of C. Nucleic Acids Res. 1990, 18, 5775–5780. [Google Scholar] [CrossRef] [PubMed]
- Focher, F.; Verri, A.; Verzeletti, S.; Mazzarello, P.; Spadari, S. Uracil in OriS of herpes simplex 1 alters its specific recognition by origin binding protein (OBP): Does virus induced uracil-DNA glycosylase play a key role in viral reactivation and replication? Chromosoma 1992, 102, S67–S71. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA glycosylases — Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014, 23, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Tye, B.K.; Nyman, P.O.; Lehman, I.R.; Hochhauser, S.; Weiss, B. Transient accumulation of Okazaki fragments as a result of uracil incorporation into nascent DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Imahashi, M.; Nakashima, M.; Iwatani, Y. Antiviral mechanism and biochemical basis of the human APOBEC3 family. Front. Microbiol. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated Uracil DNA Glycosylase-2 and Apurinic/ Apyrimidinic Endonuclease Are Involved in the Degradation of APOBEC3G-edited Nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef]
- Martomo, S.A.; Gearhart, P.J. Somatic hypermutation: Subverted DNA repair. Curr. Opin. Immunol. 2006, 18, 243–248. [Google Scholar] [CrossRef]
- Yousif, A.S.; Stanlie, A.; Mondal, S.; Honjo, T.; Begum, N.A. Differential regulation of S-region hypermutation and class-switch recombination by noncanonical functions of uracil DNA glycosylase. Proc. Natl. Acad. Sci. USA 2014, 111, E1016–E1024. [Google Scholar] [CrossRef]
- Yan, N.; O’Day, E.; Wheeler, L.A.; Engelman, A.; Lieberman, J. HIV DNA is heavily uracilated, which protects it from autointegration. Proc. Natl. Acad. Sci. USA 2011, 108, 9244–9249. [Google Scholar] [CrossRef]
- Weil, A.F.; Ghosh, D.; Zhou, Y.; Seiple, L.; McMahon, M.A.; Spivak, A.M.; Siciliano, R.F.; Stivers, J.T. Uracil DNA glycosylase initiates degradation of HIV-1 cDNA containing misincorporated dUTP and prevents viral integration. Proc. Natl. Acad. Sci. USA 2013, 110, E448–E457. [Google Scholar] [CrossRef] [PubMed]
- Hansen, E.C.; Ransom, M.; Hesselberth, J.R.; Hosmane, N.N.; Capoferri, A.A.; Bruner, K.M.; Pollack, R.A.; Zhang, H.; Drummond, M.B.; Siliciano, J.M.; et al. Diverse fates of uracilated HIV-1 DNA during infection of myeloid lineage cells. Elife 2016, 5, e18447. [Google Scholar] [CrossRef]
- Takahashi, I.; Marmur, J. Replacement of Thymidylic Acid by Deoxyuridylic Acid in the Deoxyribonucleic Acid of a Transducing Phage for Bacillus subtilis. Nature 1963, 197, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Kiljunen, S.; Hakala, K.; Pinta, E.; Huttunen, S.; Pluta, P.; Gador, A.; Lönnberg, H.; Skurnik, M. Yersiniophage ϕR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 2005, 151, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, J.; Takemura-Uchiyama, I.; Sakaguchi, Y.; Gamoh, K.; Kato, S.; Daibata, M.; Ujihara, T.; Misawa, N.; Matsuzaki, S. Intragenus generalized transduction in Staphylococcus spp. by a novel giant phage. ISME J. 2014, 8, 1949–1952. [Google Scholar] [CrossRef]
- Lavysh, D.; Sokolova, M.; Minakhin, L.; Yakunina, M.; Artamonova, T.; Kozyavkin, S.; Makarova, K.S.; Koonin, E.V.; Severinov, K. The genome of AR9, a giant transducing Bacillus phage encoding two multisubunit RNA polymerases. Virology 2016, 495, 185–196. [Google Scholar] [CrossRef]
- Duncan, B.K.; Warner, H.R. Metabolism of uracil-containing DNA: Degradation of bacteriophage PBS2 DNA in Bacillus subtilis. J. Virol. 1977, 22, 835–838. [Google Scholar] [CrossRef]
- Lindahl, T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl. Acad. Sci. USA 1974, 71, 3649–3653. [Google Scholar] [CrossRef]
- Schormann, N.; Zhukovskaya, N.; Bedwell, G.; Nuth, M.; Gillilan, R.; Prevelige, P.E.; Ricciardi, R.P.; Banerjee, S.; Chattopadhyay, D. Poxvirus uracil-DNA glycosylase-An unusual member of the family I uracil-DNA glycosylases. Protein Sci. 2016, 25, 2113–2131. [Google Scholar] [CrossRef]
- Savva, R. Targeting uracil-DNA glycosylases for therapeutic outcomes using insights from virus evolution. Future Med. Chem. 2019, 11, 1323–1344. [Google Scholar] [CrossRef]
- Pyles, R.B.; Thompson, R.L. Evidence that the herpes simplex virus type 1 uracil DNA glycosylase is required for efficient viral replication and latency in the murine nervous system. J. Virol. 1994, 68, 4963–4972. [Google Scholar] [CrossRef] [PubMed]
- Savva, R.; McAuley-Hecht, K.; Brown, T.; Pearl, L. The structural basis of specific base-excision repair by uracil-DNA glycosylase. Nature 1995, 373, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Duke, G.M.; Mocarski, E.S. Human cytomegalovirus uracil DNA glycosylase is required for the normal temporal regulation of both DNA synthesis and viral replication. J. Virol. 1996, 70, 3018–3025. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, C.T.; Courcelle, J.; Prichard, M.N.; Mocarski, E.S. Requirement for Uracil-DNA Glycosylase during the Transition to Late-Phase Cytomegalovirus DNA Replication. J. Virol. 2001, 75, 7592–7601. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Lawlor, H.; Duke, G.M.; Mo, C.; Wang, Z.; Dixon, M.; Kemble, G.; Kern, E.R. Human cytomegalovirus uracil DNA glycosylase associates with ppUL44 and accelerates the accumulation of viral DNA. Virol. J. 2005, 2, 55. [Google Scholar] [CrossRef] [PubMed]
- Ranneberg-Nilsen, T.; Rollag, H.; Slettebakk, R.; Backe, P.H.; Olsen, Ø.; Luna, L.; Bjørås, M. The chromatin remodeling factor smarcb1 forms a complex with human cytomegalovirus proteins UL114 and UL44. PLoS ONE 2012, 7, e34119. [Google Scholar] [CrossRef]
- Strang, B.L.; Coen, D.M. Interaction of the human cytomegalovirus uracil DNA glycosylase UL114 with the viral DNA polymerase catalytic subunit UL54. J. Gen. Virol. 2010, 91, 2029–2033. [Google Scholar] [CrossRef]
- Lu, C.C.; Huang, H.T.; Wang, J.T.; Slupphaug, G.; Li, T.K.; Wu, M.C.; Chen, Y.C.; Lee, C.P.; Chen, M.R. Characterization of the uracil-DNA glycosylase activity of Epstein-Barr virus BKRF3 and its role in lytic viral DNA replication. J. Virol. 2007, 81, 1195–1208. [Google Scholar] [CrossRef][Green Version]
- Su, M.T.; Liu, I.H.; Wu, C.W.; Chang, S.M.; Tsai, C.H.; Yang, P.W.; Chuang, Y.C.; Lee, C.P.; Chen, M.R. Uracil DNA glycosylase BKRF3 contributes to Epstein-Barr virus DNA replication through physical interactions with proteins in viral DNA replication complex. J. Virol. 2014, 88, 8883–8899. [Google Scholar] [CrossRef][Green Version]
- Géoui, T.; Buisson, M.; Tarbouriech, N.; Burmeister, W.P. New insights on the role of the gamma-herpesvirus uracil-DNA glycosylase leucine loop revealed by the structure of the Epstein-Barr virus enzyme in complex with an inhibitor protein. J. Mol. Biol. 2007, 366, 117–131. [Google Scholar] [CrossRef]
- Earl, C.; Bagnéris, C.; Zeman, K.; Cole, A.; Barrett, T.; Savva, R. A structurally conserved motif in γ-herpesvirus uracil-DNA glycosylases elicits duplex nucleotide-flipping. Nucleic Acids Res. 2018, 46, 4286–4300. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Maidou-Peindara, P.; Aas, P.A.; Jacquot, G.; Otterlei, M.; Slupphaug, G.; Benichou, S. Human immunodeficiency virus type 1 Vpr modulates cellular expression of UNG2 via a negative transcriptional effect. J. Virol. 2009, 83, 10256–10263. [Google Scholar] [CrossRef] [PubMed]
- Bouhamdan, M.; Benichou, S.; Rey, F.; Navarro, J.M.; Agostini, I.; Spire, B.; Camonis, J.; Slupphaug, G.; Vigne, R.; Benarous, R.; et al. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J. Virol. 1996, 70, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Eldin, P.; Chazal, N.; Fenard, D.; Bernard, E.; Guichou, J.F.; Briant, L. Vpr expression abolishes the capacity of HIV-1 infected cells to repair uracilated DNA. Nucleic Acids Res. 2013, 42, 1698–1710. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef]
- Katz, G.E.; Price, A.R.; Pomerantz, M.J. Bacteriophage PBS2-induced inhibition of uracil-containing DNA degradation. J. Virol. 1976, 20, 535–538. [Google Scholar] [CrossRef]
- Wang, Z.; Mosbaugh, D.W. UraciI-DNA glycosylase inhibitor gene of bacteriophage PBS2 encodes a binding protein specific for uraciI-DNA glycosylase. J. Biol. Chem. 1989, 264, 1163–1171. [Google Scholar]
- Warner, H.R.; Johnson, L.K.; Snustad, D.P. Early Events After Infection of Escherichia coli by Bacteriophage T5. III. Inhibition of Uracil-DNA Glycosylase Activity. J. Virol. 1980, 33, 535–538. [Google Scholar] [CrossRef]
- Serrano-Heras, G.; Bravo, A.; Salas, M. Phage φ29 protein p56 prevents viral DNA replication impairment caused by uracil excision activity of uracil-DNA glycosylase. Proc. Natl. Acad. Sci. USA 2008, 105, 19044–19049. [Google Scholar] [CrossRef]
- Yamagishi, H. Single Strand Interruptions in PBS 1 Bacteriophage DNA Molecule. J. Mol. Biol. 1968, 35, 623–633. [Google Scholar] [CrossRef]
- Rhoades, M. Localization of single-chain interruptions in bacteriophage T5 DNA. II. Electrophoretic studies. J. Virol. 1977, 23, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.S.; Krutilina, A.I.; Shlyapnikov, M.G.; Severinov, K.; Lavysh, D.; Kochetkov, V.V.; McGrath, J.W.; de Leeuwe, C.; Shaburova, O.V.; Krylov, V.N.; et al. Genomic Analysis of Pseudomonas putida Phage tf with Localized Single-Strand DNA Interruptions. PLoS ONE 2012, 7, e51163. [Google Scholar] [CrossRef] [PubMed]
- Lough, J.; Jackson, M.; Morris, R.; Moyer, R. Bisulfite-induced cytosine deamination rates in E. coli SSB:DNA complexes. Mutat. Res. 2001, 478, 191–197. [Google Scholar] [CrossRef]
- Conticello, S.G.; Langlois, M.A.; Yang, Z.; Neuberger, M.S. DNA deamination in immunity: AID in the context of its APOBEC relatives. Adv. Immunol. 2007, 94, 37–73. [Google Scholar] [CrossRef]
- Silvas, T.V.; Schiffer, C.A. APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci. 2019, 28, 1552–1566. [Google Scholar] [CrossRef]
- Pautasso, S.; Galitska, G.; Dell’Oste, V.; Biolatti, M.; Cagliani, R.; Forni, D.; De Andrea, M.; Gariglio, M.; Sironi, M.; Landolfo, S. Strategy of Human Cytomegalovirus To Escape Interferon Beta-Induced APOBEC3G Editing Activity. J. Virol. 2018, 92, e01224-18. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E.; et al. Epstein–Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat. Microbiol 2019, 4, 78–88. [Google Scholar] [CrossRef]
- Verma, S.C.; Bajaj, B.G.; Cai, Q.; Si, H.; Seelhammer, T.; Robertson, E.S. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus recruits uracil DNA glycosylase 2 at the terminal repeats and is important for latent persistence of the virus. J. Virol. 2006, 80, 11178–11190. [Google Scholar] [CrossRef][Green Version]
- Cole, A.R.; Ofer, S.; Ryzhenkova, K.; Baltulionis, G.; Hornyak, P.; Savva, R. Architecturally diverse proteins converge on an analogous mechanism to inactivate Uracil-DNA glycosylase. Nucleic Acids Res. 2013, 41, 8760–8775. [Google Scholar] [CrossRef][Green Version]
- Wang, H.C.; Hsu, K.C.; Yang, J.M.; Wu, M.L.; Ko, T.P.; Lin, S.R.; Wang, A.H. Staphylococcus aureus protein SAUGI acts as a uracil-DNA glycosylase inhibitor. Nucleic Acids Res. 2014, 42, 1354–1364. [Google Scholar] [CrossRef][Green Version]
- Wu, Y.; Zhou, X.; Barnes, C.O.; DeLucia, M.; Cohen, A.E.; Gronenborn, A.M.; Ahn, J.; Calero, G. The DDB1–DCAF1–Vpr–UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Von Hippel, P.H.; Johnson, N.P.; Marcus, A.H. Fifty years of DNA “Breathing”: Reflections on old and new approaches. Biopolymers 2013, 99, 923–954. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Mol, C.D.; Slupphaug, G.; Bharati, S.; Krokan, H.E.; Tainer, J.A. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998, 17, 5214–5226. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.B.; Bianchet, M.A.; Krosky, D.J.; Friedman, J.I.; Amzel, L.M.; Stivers, J.T. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature 2007, 449, 433–437. [Google Scholar] [CrossRef]
- Wang, H.C.; Ho, C.H.; Chou, C.C.; Ko, T.P.; Huang, M.F.; Hsu, K.C.; Wang, A.H. Using structural-based protein engineering to modulate the differential inhibition effects of SAUGI on human and HSV uracil DNA glycosylase. Nucleic Acids Res. 2016, 44, 4440–4449. [Google Scholar] [CrossRef]
- Minkah, N.; Macaluso, M.; Oldenburg, D.G.; Paden, C.R.; White, D.W.; McBride, K.M.; Krug, L.T. Absence of the uracil DNA glycosylase of murine gammaherpesvirus 68 impairs replication and delays the establishment of latency in vivo. J. Virol. 2015, 89, 3366–3379. [Google Scholar] [CrossRef][Green Version]
- Mullaney, J.; Moss, H.W.; McGeoch, D.J. Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase. J. Gen. Virol. 1989, 70, 449–454. [Google Scholar] [CrossRef]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the Herpes Simplex Virus-1 DNA Polymerase and Uracil DNA Glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef]
- Hernández Durán, A.; Grünewald, K.; Topf, M. Conserved Central Intraviral Protein Interactome of the Herpesviridae Family. mSystems 2019, 4, e00295-19. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Sheng, X.; Murray-Nerger, L.A.; Cristea, I.M. Temporal dynamics of protein complex formation and dissociation during human cytomegalovirus infection. Nat. Commun. 2020, 11, 806. [Google Scholar] [CrossRef]
- Argnani, R.; Focher, F.; Zucchini, S.; Verri, A.; Wright, G.E.; Spadari, S.; Manservigi, R. Herpes simplex virus type 1 (HSV-1) uracil-DNA glycosylase: Functional expression in Escherichia coli, biochemical characterization, and selective inhibition by 6-(p-n-octylanilino)uracil. Virology 1995, 211, 307–311. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, H.; Zhi, C.; Wright, G.E.; Ubiali, D.; Pregnolato, M.; Verri, A.; Focher, F.; Spadari, S. Molecular Modeling and Synthesis of Inhibitors of Herpes Simplex Virus Type 1 Uracil-DNA Glycosylase. J. Med. Chem. 1999, 42, 2344–2350. [Google Scholar] [CrossRef] [PubMed]
- Sekino, Y.; Bruner, S.D.; Verdine, G.L. Selective inhibition of herpes simplex virus type-1 uracil-DNA glycosylase by designed substrate analogs. J. Biol. Chem. 2000, 275, 36506–36508. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.L.; Krosky, D.J.; Seiple, L.; Stivers, J.T. Uracil-directed ligand tethering: An efficient strategy for uracil DNA glycosylase (UNG) inhibitor development. J. Am. Chem. Soc. 2005, 127, 17412–17420. [Google Scholar] [CrossRef]
- Hendricks, U.; Crous, W.; Naidoo, K.J. Computational Rationale for the Selective Inhibition of the Herpes Simplex Virus Type 1 Uracil-DNA Glycosylase Enzyme. J. Chem. Inf. Model. 2014, 54, 3362–3372. [Google Scholar] [CrossRef]
- Dong, Q.; Smith, K.R.; Oldenburg, D.G.; Shapiro, M.; Schutt, W.R.; Malik, L.; Plummer, J.B.; Mu, Y.; MacCarthy, T.; White, D.W.; et al. Combinatorial Loss of the Enzymatic Activities of Viral Uracil-DNA Glycosylase and Viral dUTPase Impairs Murine Gammaherpesvirus Pathogenesis and Leads to Increased Recombination-Based Deletion in the Viral Genome. mBio 2018, 9, e01831-18. [Google Scholar] [CrossRef]
- Krosky, D.J.; Bianchet, M.A.; Seiple, L.; Chung, S.; Amzel, L.M.; Stivers, J.T. Mimicking damaged DNA with a small molecule inhibitor of human UNG2. Nucleic Acids Res. 2006, 34, 5872–5879. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Chung, S.; Krosky, D.J.; Stivers, J.T. Synthesis and high-throughput evaluation of triskelion uracil libraries for inhibition of human dUTPase and UNG2. Bioorg. Med. Chem. 2006, 14, 5666–5672. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Common Name | Virus Numeric Name | Herpesviridae Subfamily | Reference Genome Accession Code | Genome Length | Uracil-DNA Glycosylase Accession Code |
|---|---|---|---|---|---|
| HSV-1 | HHV-1 | Alphaherpesvirinae | NC_001806 | 152222 | YP_009137076 |
| HSV-2 | HHV-2 | NC_001798 | 154675 | YP_009137153 | |
| VZV | HHV-3 | NC_001348 | 124884 | NP_040181 | |
| HCMV | HHV-5 | Betaherpesvirinae | NC_006273 | 235646 | YP_081554 |
| - | HHV-6A | NC_001664 | 159378 | NP_042974 | |
| - | HHV-6B | NC_000898 | 162114 | NP_050260 | |
| - | HHV-7 | NC_001716 | 153080 | YP_073819 | |
| EBV | HHV-4 ** | Gammaherpesvirinae | NC_007605 | 171823 | YP_401679 |
| KSHV | HHV-8 | NC_009333 | 137969 | YP_001129398 |
| Canonical State | Protective Factors | Post-Error State | Consequence of Error | Repair Measure |
|---|---|---|---|---|
| C:G base-pair | dsDNA state or protein complex | U:G mistmatch | promutagenic | UDG |
| 5-me C:G base-pair | dsDNA state or protein complex | T:G mismatch | promutagenic | TDG |
| T:A base-pair | dUTPase and DNA polymerase | U:A base-pair | dysregulatory | UDG |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savva, R. The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design. Microorganisms 2020, 8, 461. https://doi.org/10.3390/microorganisms8030461
Savva R. The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design. Microorganisms. 2020; 8(3):461. https://doi.org/10.3390/microorganisms8030461
Chicago/Turabian StyleSavva, Renos. 2020. "The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design" Microorganisms 8, no. 3: 461. https://doi.org/10.3390/microorganisms8030461
APA StyleSavva, R. (2020). The Essential Co-Option of Uracil-DNA Glycosylases by Herpesviruses Invites Novel Antiviral Design. Microorganisms, 8(3), 461. https://doi.org/10.3390/microorganisms8030461