Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression

 ,
,  ,
,  , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
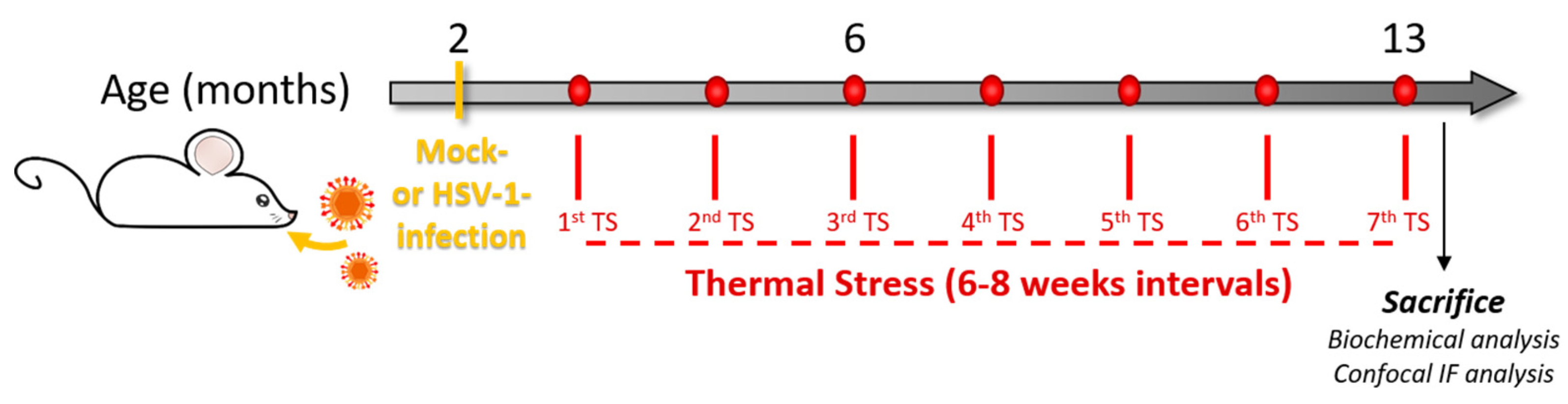
2.2. Mouse Model of Recurrent HSV-1 Infection
2.3. Western Blotting
2.4. 2D Electrophoresis
2.5. 2D Western Blot
2.6. Image Analysis
2.7. In-gel Trypsin Digestion/Peptide Extraction
2.8. Reverse Phase -HPLC-High Resolution MS/MS Characterization of Tryptic Peptides
2.9. MS Data Analysis
2.10. Immunoprecipitation
2.11. Immunofluorescence and Confocal Microscopy
2.12. Statistics
3. Results
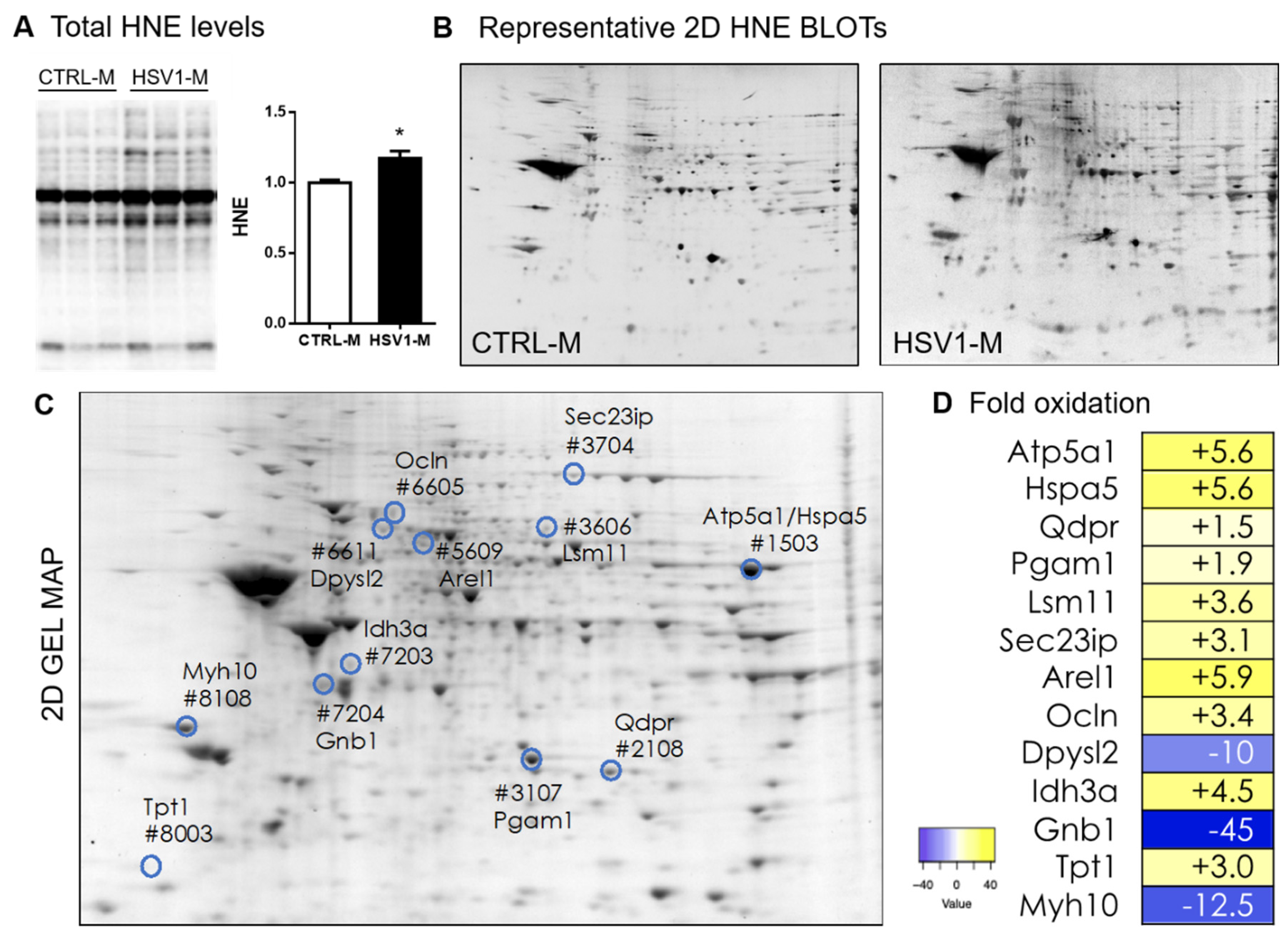
3.1. Recurrent HSV-1 Infection Induces Oxidative Modifications of Proteins in Mouse Cortices
3.2. Recurrent HSV-1 Infection Induces an Altered HNE Profile in Mouse Cortices
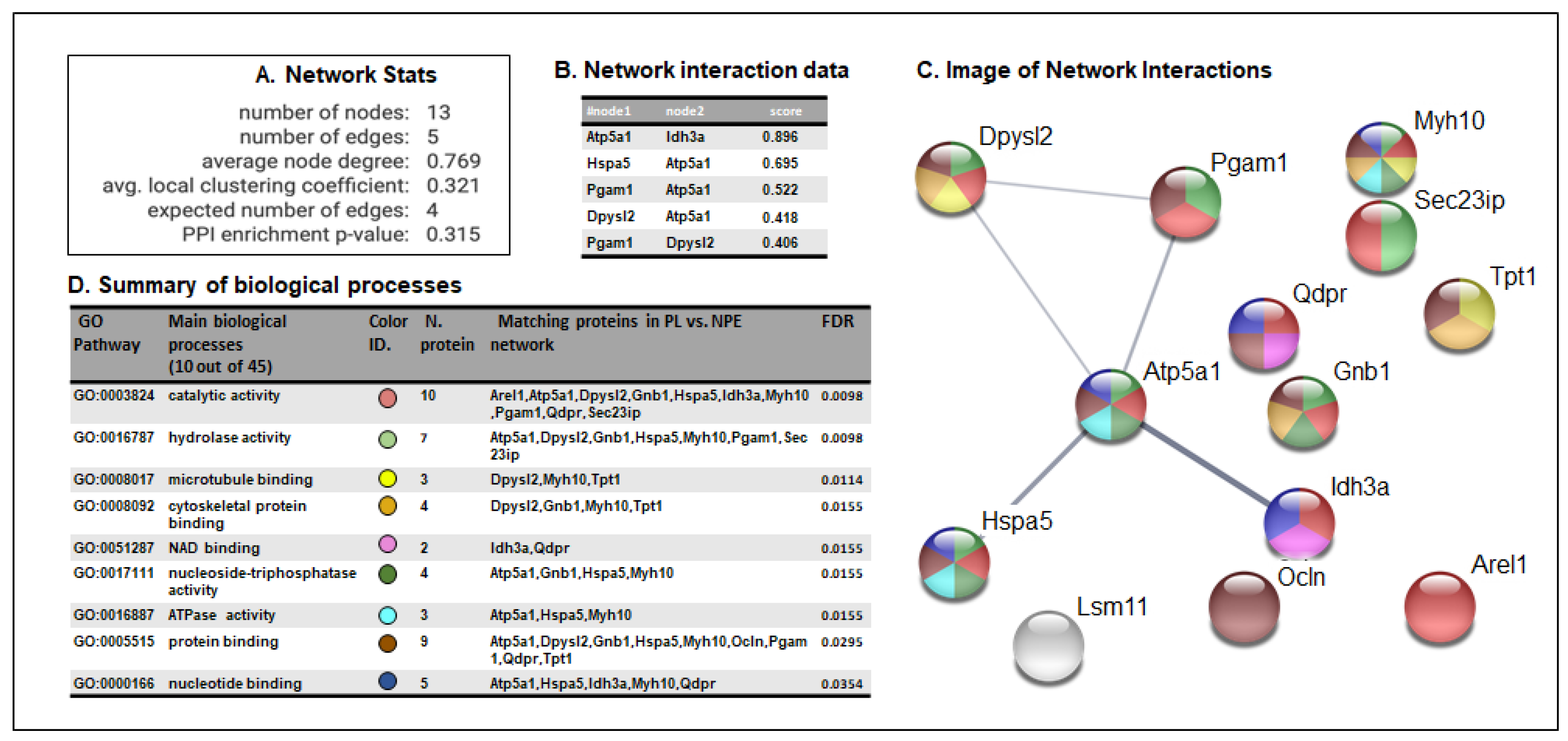
3.3. STRING Analysis of Protein Networks
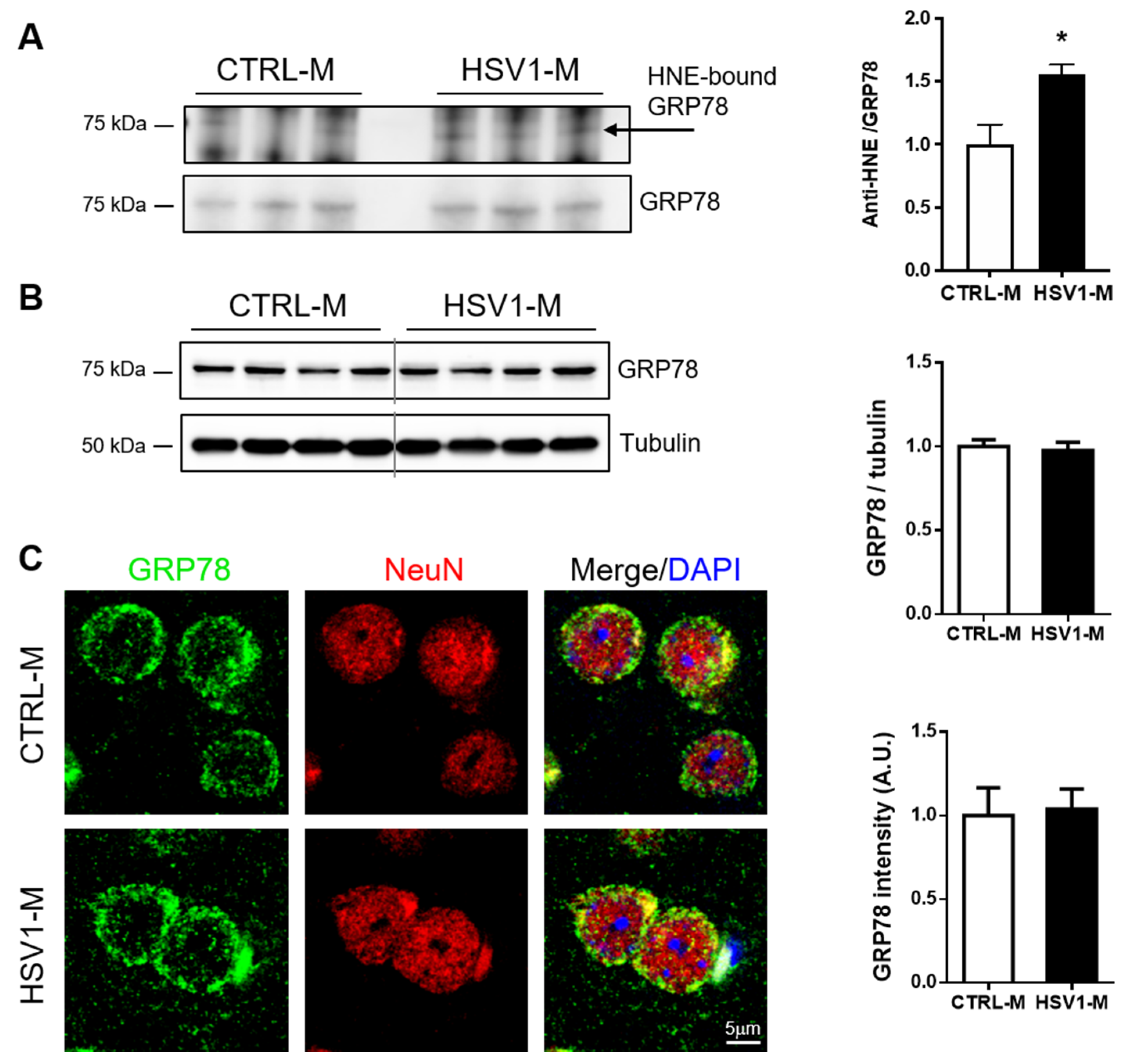
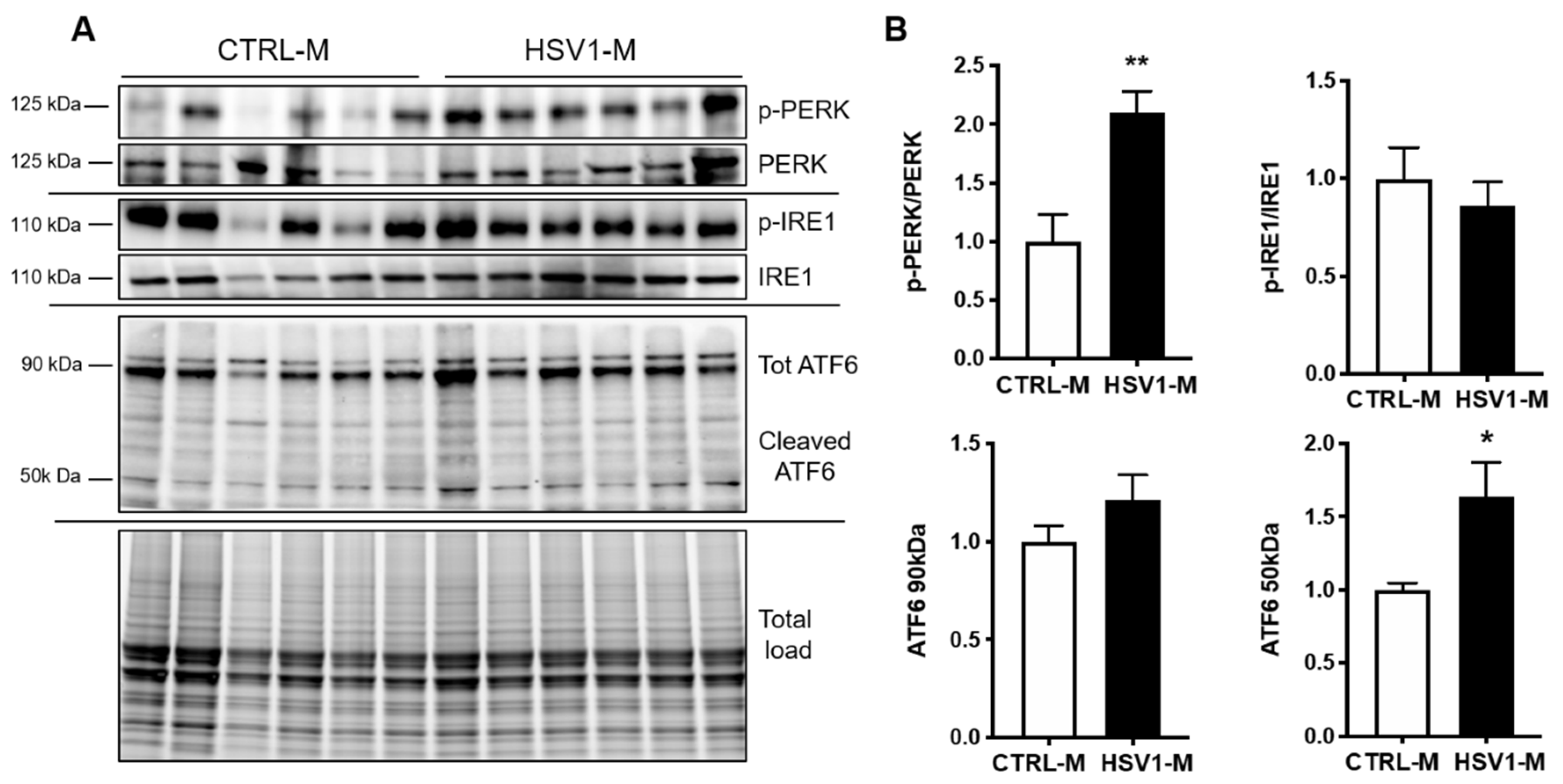
3.4. GRP78 Focus
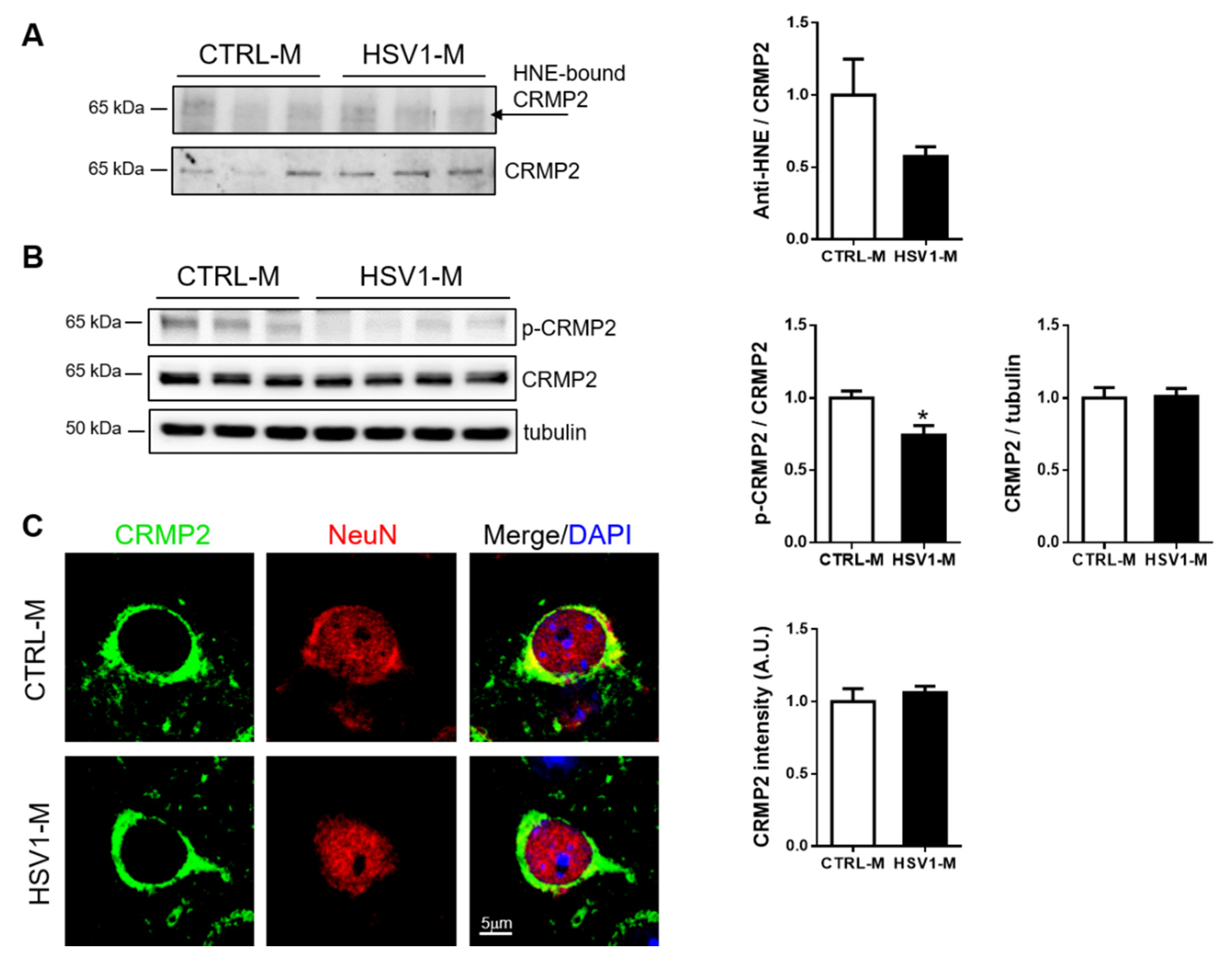
3.5. CRMP2 Focus
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious agents and neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef]
- Harris, S.A.; Harris, E.A. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2015, 48, 319–353. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Itzhaki, R. Herpes simplex virus type 1, apolipoprotein E and Alzheimer’ disease. Herpes 2004, 11, 77A–82A. [Google Scholar]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef]
- Letenneur, L.; Peres, K.; Fleury, H.; Garrigue, I.; Barberger-Gateau, P.; Helmer, C.; Orgogozo, J.M.; Gauthier, S.; Dartigues, J.F. Seropositivity to herpes simplex virus antibodies and risk of Alzheimer’s disease: A population-based cohort study. PLoS ONE 2008, 3, e3637. [Google Scholar] [CrossRef]
- Lovheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 593–599. [Google Scholar] [CrossRef]
- Kobayashi, N.; Nagata, T.; Shinagawa, S.; Oka, N.; Shimada, K.; Shimizu, A.; Tatebayashi, Y.; Yamada, H.; Nakayama, K.; Kondo, K. Increase in the IgG avidity index due to herpes simplex virus type 1 reactivation and its relationship with cognitive function in amnestic mild cognitive impairment and Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2013, 430, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Baglio, F.; Cabinio, M.; Calabrese, E.; Hernis, A.; Nemni, R.; Clerici, M. Titers of herpes simplex virus type 1 antibodies positively correlate with grey matter volumes in Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Lopatko Lindman, K.; Weidung, B.; Olsson, J.; Josefsson, M.; Kok, E.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F.; Lovheim, H. A genetic signature including apolipoprotein Eepsilon4 potentiates the risk of herpes simplex-associated Alzheimer’s disease. Alzheimer’s Dement. 2019, 5, 697–704. [Google Scholar]
- Linard, M.; Letenneur, L.; Garrigue, I.; Doize, A.; Dartigues, J.F.; Helmer, C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 200–208. [Google Scholar] [CrossRef]
- Shipley, S.J.; Parkin, E.T.; Itzhaki, R.F.; Dobson, C.B. Herpes simplex virus interferes with amyloid precursor protein processing. BMC Microbiol. 2005, 5, 48. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Itzhaki, R.F.; Shipley, S.J.; Dobson, C.B. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci. Lett. 2007, 429, 95–100. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef]
- Piacentini, R.; Civitelli, L.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Azzena, G.B.; Palamara, A.T.; Grassi, C. HSV-1 promotes Ca2+ -mediated APP phosphorylation and Abeta accumulation in rat cortical neurons. Neurobiol. Aging 2011, 32, 2323.e13–26. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Palsson, S.; Roberts, T.C.; Jarver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levanen, B.; Skold, M.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Alvarez, G.; Aldudo, J.; Alonso, M.; Santana, S.; Valdivieso, F. Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J. Neurosci. Res. 2012, 90, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, R.; Li Puma, D.D.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Palamara, A.T.; Grassi, C. Herpes simplex virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-beta protein accumulation. Sci. Rep. 2015, 5, 15444. [Google Scholar] [CrossRef] [PubMed]
- Civitelli, L.; Marcocci, M.E.; Celestino, I.; Piacentini, R.; Garaci, E.; Grassi, C.; De Chiara, G.; Palamara, A.T. Herpes simplex virus type 1 infection in neurons leads to production and nuclear localization of APP intracellular domain (AICD): Implications for Alzheimer’s disease pathogenesis. J. Neurovirol. 2015, 21, 480–490. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Racaniello, M.; Mollinari, C.; Marcocci, M.E.; Aversa, G.; Cardinale, A.; Giovanetti, A.; Garaci, E.; Palamara, A.T.; Merlo, D. Herpes simplex virus-type1 (HSV-1) impairs DNA repair in cortical neurons. Front. Aging Neurosci. 2016, 8, 242. [Google Scholar] [CrossRef]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef]
- Li Puma, D.D.; Piacentini, R.; Leone, L.; Gironi, K.; Marcocci, M.E.; De Chiara, G.; Palamara, A.T.; Grassi, C. Herpes simplex virus type-1 infection impairs adult hippocampal neurogenesis via amyloid-beta protein accumulation. Stem Cells 2019, 37, 1467–1480. [Google Scholar] [CrossRef]
- Nucci, C.; Palamara, A.T.; Ciriolo, M.R.; Nencioni, L.; Savini, P.; D’Agostini, C.; Rotilio, G.; Cerulli, L.; Garaci, E. Imbalance in corneal redox state during herpes simplex virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp. Eye Res. 2000, 70, 215–220. [Google Scholar] [CrossRef]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol. 2007, 13, 416–425. [Google Scholar] [CrossRef]
- Valyi-Nagy, T.; Dermody, T.S. Role of oxidative damage in the pathogenesis of viral infections of the nervous system. Histol. Histopathol. 2005, 20, 957–967. [Google Scholar]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Moutal, A.; White, K.A.; Chefdeville, A.; Laufmann, R.N.; Vitiello, P.F.; Feinstein, D.; Weimer, J.M.; Khanna, R. Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Mol. Neurobiol. 2019, 56, 6736–6755. [Google Scholar] [CrossRef] [PubMed]
- Sawtell, N.M.; Thompson, R.L. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J. Virol. 1992, 66, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Smith, M.A. Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 2008, 21, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Hardas, S.S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; De Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Bibioophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef]
- Lanzillotta, C.; Tramutola, A.; Meier, S.; Schmitt, F.; Barone, E.; Perluigi, M.; Di Domenico, F.; Abisambra, J.F. Early and selective activation and subsequent alterations to the unfolded protein response in down syndrome mouse models. J. Alzheimer’s Dis. 2018, 62, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: An emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol. 2011, 43, 180–191. [Google Scholar] [CrossRef]
- Uchida, Y.; Goshima, Y. Molecular mechanism of axon guidance mediated by phosphorylation of CRMP2. Seikagaku 2005, 77, 1424–1427. [Google Scholar] [PubMed]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes simplex virus-1 in the brain: The dark side of a sneaky infection. Trends Microbiol. 2020, S0966-842X(20)30074-3. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural. Regen. Res. 2012, 7, 376–385. [Google Scholar] [PubMed]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Sultana, R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 2011, 45, 59–72. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta Neuropathol. 2009, 118, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Sultana, R.; Cenini, G.; Di Domenico, F.; Memo, M.; Pierce, W.M.; Coccia, R.; Butterfield, D.A. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteom. Clin. Appl. 2009, 3, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Perluigi, M.; Newman, S.F.; Pierce, W.M.; Cini, C.; Coccia, R.; Butterfield, D.A. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxid. Redox Signal. 2010, 12, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Fiorini, A.; Sultana, R.; Perluigi, M.; Cai, J.; Klein, J.B.; Head, E.; Butterfield, D.A. An investigation of the molecular mechanisms engaged before and after the development of Alzheimer disease neuropathology in Down syndrome: A proteomics approach. Free Radic. Biol. Med. 2014, 76, 89–95. [Google Scholar] [CrossRef]
- Yang, Y.; Turner, R.S.; Gaut, J.R. The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J. Biol. Chem. 1998, 273, 25552–25555. [Google Scholar] [CrossRef]
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and coordinator of organismal homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; Group, P.S.P.G.S.; Lee, V.M.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef]
- Hamos, J.E.; Oblas, B.; Pulaski-Salo, D.; Welch, W.J.; Bole, D.G.; Drachman, D.A. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991, 41, 345–350. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Kakimura, J.; Kitamura, Y.; Taniguchi, T.; Shimohama, S.; Gebicke-Haerter, P.J. Bip/GRP78-induced production of cytokines and uptake of amyloid-beta(1-42) peptide in microglia. Biochem. Biophys. Res. Commun. 2001, 281, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Tsuchiya, D.; Taniguchi, T.; Gebicke-Haerter, P.J.; Smith, M.A.; Perry, G.; Shimohama, S. Possible involvement of ER chaperone Grp78 on reduced formation of amyloid-beta deposits. Ann. N. Y. Acad. Sci. 2002, 977, 327–332. [Google Scholar] [CrossRef]
- Endres, K.; Reinhardt, S. ER-stress in Alzheimer’s disease: Turning the scale? Am. J. Neurodegener Dis. 2013, 2, 247–265. [Google Scholar] [PubMed]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Liu, Z.C.; Fu, Z.Q.; Song, J.; Zhang, J.Y.; Wei, Y.P.; Chu, J.; Han, L.; Qu, N.; Wang, J.Z.; Tian, Q. Bip enhanced the association of GSK-3beta with tau during ER stress both in vivo and in vitro. J. Alzheimer’s Dis. 2012, 29, 727–740. [Google Scholar] [CrossRef]
- Liu, Z.C.; Chu, J.; Lin, L.; Song, J.; Ning, L.N.; Luo, H.B.; Yang, S.S.; Shi, Y.; Wang, Q.; Qu, N.; et al. SIL1 rescued bip elevation-related tau hyperphosphorylation in ER stress. Mol. Neurobiol. 2016, 53, 983–994. [Google Scholar] [CrossRef]
- Ha, Y.; Liu, W.; Liu, H.; Zhu, S.; Xia, F.; Gerson, J.E.; Azhar, N.A.; Tilton, R.G.; Motamedi, M.; Kayed, R.; et al. AAV2-mediated GRP78 transfer alleviates retinal neuronal injury by downregulating ER stress and tau oligomer formation. Invest. Ophthalmol. Vis. Sci. 2018, 59, 4670–4682. [Google Scholar] [CrossRef]
- Di Domenico, F.; Coccia, R.; Cocciolo, A.; Murphy, M.P.; Cenini, G.; Head, E.; Butterfield, D.A.; Giorgi, A.; Schinina, M.E.; Mancuso, C.; et al. Impairment of proteostasis network in down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim. Biophys. Acta 2013, 1832, 1249–1259. [Google Scholar] [CrossRef]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Palmer, D.; Rosenthal, K.S. Changes in BiP (GRP78) levels upon HSV-1 infection are strain dependent. Virus Res. 2001, 76, 127–135. [Google Scholar] [CrossRef]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Su, A.; Wang, H.; Li, Y.; Wang, X.; Chen, D.; Wu, Z. Opposite roles of RNase and kinase activities of inositol-requiring enzyme 1 (IRE1) on HSV-1 replication. Viruses 2017, 9, 235. [Google Scholar] [CrossRef]
- Moller, D.; Gellert, M.; Langel, W.; Lillig, C.H. Molecular dynamics simulations and in vitro analysis of the CRMP2 thiol switch. Mol. Biosyst. 2017, 13, 1744–1753. [Google Scholar] [CrossRef]
- Morinaka, A.; Yamada, M.; Itofusa, R.; Funato, Y.; Yoshimura, Y.; Nakamura, F.; Yoshimura, T.; Kaibuchi, K.; Goshima, Y.; Hoshino, M.; et al. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 2011, 4, ra26. [Google Scholar] [CrossRef]
- Soutar, M.P.; Thornhill, P.; Cole, A.R.; Sutherland, C. Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr. Alzheimer Res. 2009, 6, 269–278. [Google Scholar] [CrossRef]
- Hensley, K.; Kursula, P. Collapsin response mediator protein-2 (CRMP2) is a plausible etiological factor and potential therapeutic target in Alzheimer’s disease: Comparison and contrast with microtubule-associated protein Tau. J. Alzheimer’s Dis. 2016, 53, 1–14. [Google Scholar] [CrossRef]
- Martin, C.; Aguila, B.; Araya, P.; Vio, K.; Valdivia, S.; Zambrano, A.; Concha, M.I.; Otth, C. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J. Alzheimer’s Dis. 2014, 39, 849–859. [Google Scholar] [CrossRef]
- Kimura, T.; Watanabe, H.; Iwamatsu, A.; Kaibuchi, K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J. Neurochem. 2005, 93, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Hattori, A.; Kimura, T.; Nakamuta, S.; Funahashi, Y.; Hirotsune, S.; Furuta, K.; Urano, T.; Toyoshima, Y.Y.; Kaibuchi, K. CRMP-2 directly binds to cytoplasmic dynein and interferes with its activity. J. Neurochem. 2009, 111, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B. Herpes simplex viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002, 82, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.; Poon, H.F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schinina, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spot N° | Protein | Uniprot N° | Fold | Score | Coverage | Peptide | Mw | pI |
|---|---|---|---|---|---|---|---|---|
| 1503 | ATP synthase subunit alpha, mitochondrial OS = Mus musculus GN = Atp5a1 PE = 1 SV = 1 - [ATPA_MOUSE] | Q03265 | 5.6↑ | 24 | 21.7 | 11 | 59.7 | 9.19 |
| 78 kDa glucose-regulated protein OS = Mus musculus GN = Hspa5 PE = 1 SV = 3 - [GRP78_MOUSE] | P20029 | 2.1 | 2.4 | 1 | 72.4 | 5.16 | ||
| 2108 | Dihydropteridine reductase OS = Mus musculus GN = Qdpr PE = 1 SV = 2 - [DHPR_MOUSE] | Q8BVI4 | 1.5↑ | 4.75 | 18.67 | 3 | 25.6 | 78.1 |
| 3107 | Phosphoglycerate mutase 1 OS = Mus musculus GN = Pgam1 PE = 1 SV = 3 - [PGAM1_MOUSE] | Q9DBJ1 | 1.9↑ | 6.58 | 21.26 | 4 | 28.8 | 7.81 |
| 3606 | U7 snRNA-associated Sm-like protein LSm11 OS = Mus musculus GN = Lsm11 PE = 1 SV = 1 - [LSM11_MOUSE] | Q8BUV6 | 3.6↑ | 2 | 6.93 | 1 | 39.9 | 10.73 |
| 3704 | SEC23-interacting protein OS = Mus musculus GN = Sec23ip PE = 1 SV = 2 - [S23IP_MOUSE] | Q6NZC7 | 3.1↑ | 2.66 | 2.1 | 1 | 110.7 | 5.94 |
| 5609 | Apoptosis-resistant E3 ubiquitin protein ligase 1 OS = Mus musculus GN = Arel1 PE = 2 SV = 2 - [AREL1_MOUSE] | Q8CHG5 | 5.9↑ | 2.41 | 1.58 | 1 | 94.1 | 7.25 |
| 6605 | Occludin OS=Mus musculus GN = Ocln PE = 1 SV = 1 - [OCLN_MOUSE] | Q61146 | 3.4↑ | 2.26 | 1.73 | 1 | 59 | 6.48 |
| 6611 | Dihydropyrimidinase-related protein 2 OS = Mus musculus GN = Dpysl2 PE = 1 SV = 2 - [DPYL2_MOUSE] | O08553 | 10.0↓ | 2.78 | 1.75 | 1 | 62.2 | 6.36 |
| 7203 | Isocitrate dehydrogenase [NAD] subunit alpha, mitochondrial OS = Mus musculus GN = Idh3a PE = 1 SV = 1 - [IDH3A_MOUSE] | Q9D6R2 | 4.5↑ | 5.6 | 9.84 | 3 | 39.6 | 6.73 |
| 7204 | Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-1 OS=Mus musculus GN = Gnb1 PE = 1 SV = 3 - [GBB1_MOUSE] | P62874 | 45↓ | 4.38 | 7.35 | 3 | 37.4 | 6 |
| 8003 | Translationally-controlled tumor protein OS=Mus musculus GN = Tpt1 PE = 1 SV = 1 - [TCTP_MOUSE] | P63028 | 3.0↑ | 2.19 | 7.56 | 1 | 19.4 | 4.86 |
| 8108 | Myosin-10 OS = Mus musculus GN = Myh10 PE = 1 SV = 2 - [MYH10_MOUSE] | Q61879 | 12.5↓ | 2.08 | 0.4 | 1 | 228 | 5.54 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Protto, V.; Tramutola, A.; Fabiani, M.; Marcocci, M.E.; Napoletani, G.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Perluigi, M.; Di Domenico, F.; et al. Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms 2020, 8, 972. https://doi.org/10.3390/microorganisms8070972
Protto V, Tramutola A, Fabiani M, Marcocci ME, Napoletani G, Iavarone F, Vincenzoni F, Castagnola M, Perluigi M, Di Domenico F, et al. Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms. 2020; 8(7):972. https://doi.org/10.3390/microorganisms8070972
Chicago/Turabian StyleProtto, Virginia, Antonella Tramutola, Marco Fabiani, Maria Elena Marcocci, Giorgia Napoletani, Federica Iavarone, Federica Vincenzoni, Massimo Castagnola, Marzia Perluigi, Fabio Di Domenico, and et al. 2020. "Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression" Microorganisms 8, no. 7: 972. https://doi.org/10.3390/microorganisms8070972
APA StyleProtto, V., Tramutola, A., Fabiani, M., Marcocci, M. E., Napoletani, G., Iavarone, F., Vincenzoni, F., Castagnola, M., Perluigi, M., Di Domenico, F., De Chiara, G., & Palamara, A. T. (2020). Multiple Herpes Simplex Virus-1 (HSV-1) Reactivations Induce Protein Oxidative Damage in Mouse Brain: Novel Mechanisms for Alzheimer’s Disease Progression. Microorganisms, 8(7), 972. https://doi.org/10.3390/microorganisms8070972