Complete Genome Sequence Reveals Evolutionary and Comparative Genomic Features of Xanthomonas albilineans Causing Sugarcane Leaf Scald

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of Bacteria and DNA Preparation
2.2. Genome Sequencing and Assembly
2.3. Genome Component Prediction and Gene Annotation
2.4. Average Nucleotide Identity and Phylogenetic Analysis
2.5. Comparative Genomic Analysis
3. Results
3.1. General Genomic Features of X. albilineans Strain Xa-FJ1
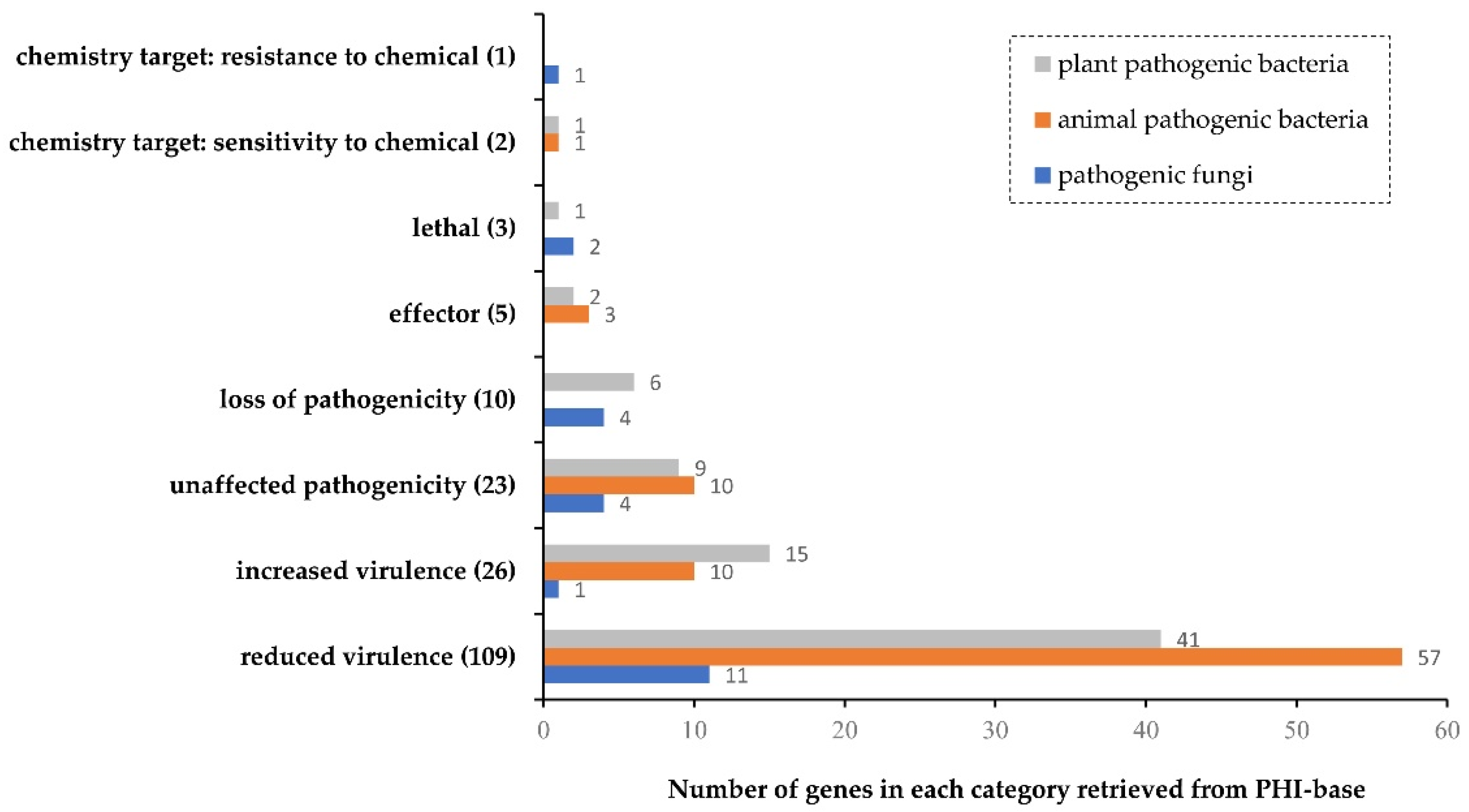
3.2. Functional Annotation of the Predicted Genes of X. albilineans Strain Xa-FJ1
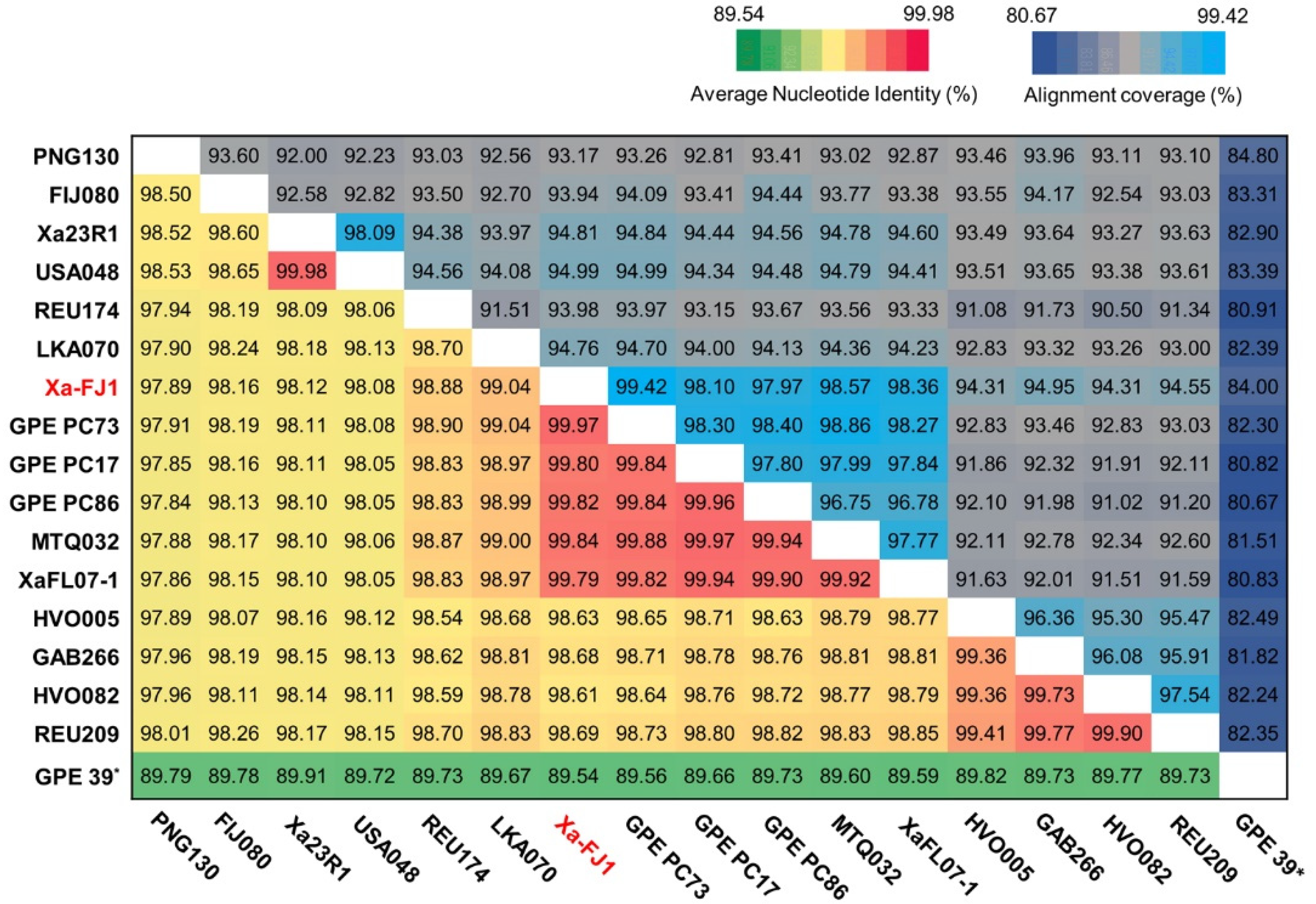
3.3. Average Nucleotide Identity and Phylogenetic Analysis among Strains of X. albilineans
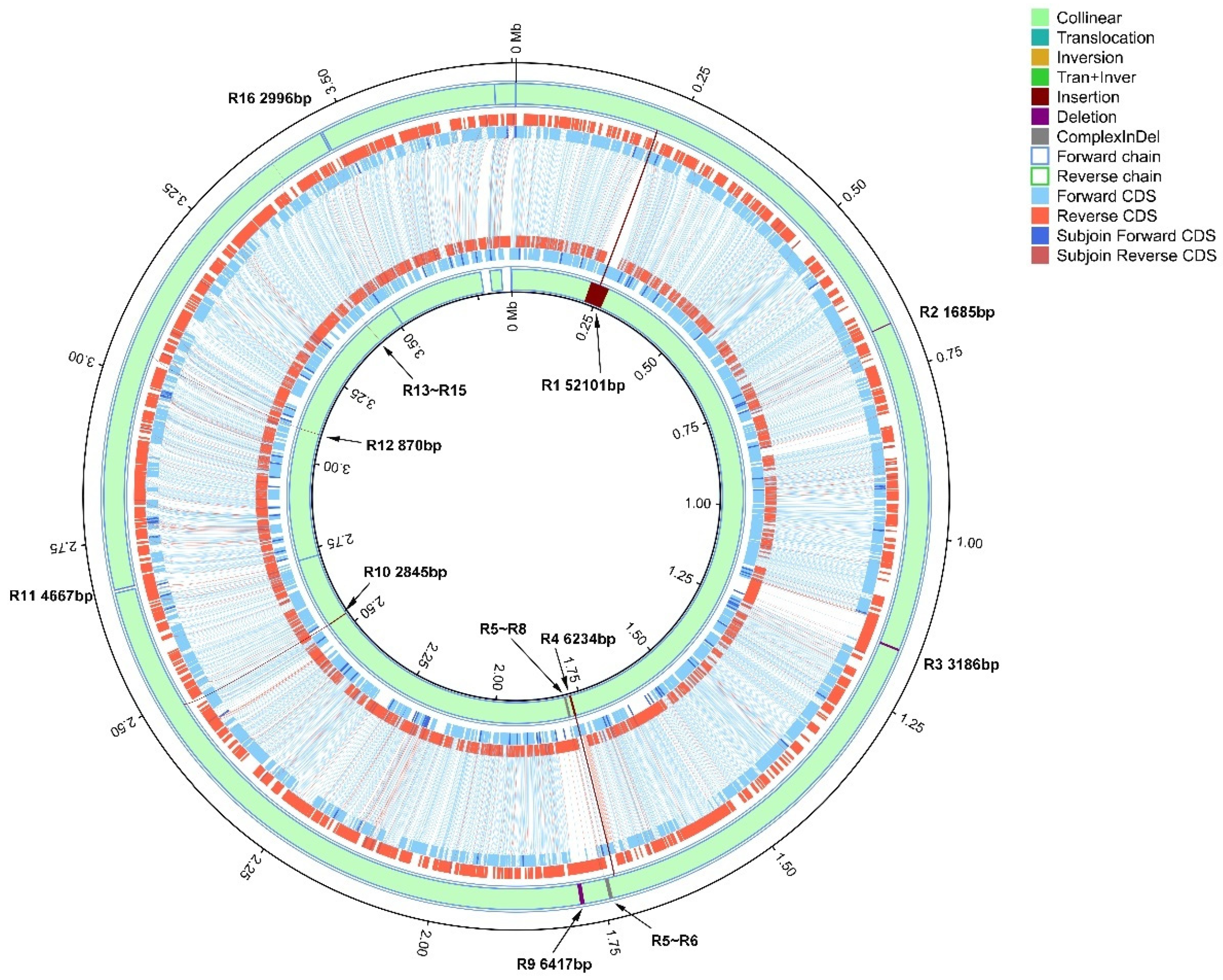
3.4. Chromosomal Structural Variation Between Strains Xa-FJ1 and GPE PC73 of X. albilineans
3.5. Chromosomal Specific Genes between Strain Xa-FJ1 and Strain GPE PC73 of X. albilineans
3.6. Genes Present in Plasmids, PlasmI and PlasmIII, of Strain GPE PC73 of X. albilineans from Guadeloupe and Absent in Strain Xa-FJ1 from China
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meyer, D.F.; Bogdanove, A.J. Genomics-driven advances in Xanthomonas biology. In Plant Pathogenic Bacteria: Genomics and Molecular Biology; Jackson, R.W., Ed.; Caister Academic Press: Norfolk, UK, 2009; pp. 147–161. [Google Scholar]
- Rott, P.; Davis, M.J. Leaf scald. In A Guide to Sugarcane Diseases; Rott, P., Bailey, R.A., Comstock, J.C., Croft, B.J., Saumtally, S., Eds.; CIRAD/ISSCT: Montpellier, France, 2000; pp. 38–44. [Google Scholar]
- Mensi, I.; Vernerey, M.S.; Gargani, D.; Nicole, M.; Rott, P. Breaking dogmas: The plant vascular pathogen Xanthomonas albilineans is able to invade non-vascular tissues despite its reduced genome. Open Biol. 2014, 4, 130116. [Google Scholar] [CrossRef] [PubMed]
- Rott, P.; Mohamed, I.S.; Klett, P.; Soupa, D.; de Saint-Albin, A.; Feldmann, P.; Letourmy, P. Resistance to leaf scald disease is associated with limited colonization of sugarcane and wild relatives by Xanthomonas albilineans. Phytopathology 1997, 87, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Rott, P.; Marguerettaz, M.; Fleites, L.; Cociancich, S.; Girard, J.-C.; Pieretti, I.; Gabriel, D.W.; Royer, M. Unravelling pathogenicity of Xanthomonas albilineans, the causal agent of sugarcane leaf scald. In Proceedings of the 27th Congress of the International Society of Sugarcane Technologists, Veracruz, Mexico, 7–11 March 2010; pp. 1112–1121. [Google Scholar]
- Hashimi, S.M.; Wall, M.K.; Smith, A.B.; Maxwell, A.; Birch, R.G. The phytotoxin albicidin is a novel inhibitor of DNA gyrase. Antimicrob. Agents Chemother. 2007, 51, 181–187. [Google Scholar] [CrossRef]
- Rott, P.; Arnaud, M.; Baudin, P. Serological and lysotypical variability of Xanthomonas albilineans (Ashby) Dowson, causal agent of sugarcane leaf scald disease. J. Phytopathol. 1986, 116, 201–211. [Google Scholar] [CrossRef]
- Rott, P.; Davis, M.J.; Baudin, P. Serological variability in Xanthomonas albilineans, causal agent of leaf scald disease of sugarcane. Plant Pathol. 1994, 43, 344–349. [Google Scholar] [CrossRef]
- Alvarez, A.; Schenck, S.; Benedict, A. Differentiation of Xanthomonas albilineans strains with monoclonal antibody reaction patterns and DNA fingerprints. Plant Pathol. 1996, 45, 358–366. [Google Scholar] [CrossRef]
- Davis, M.J.; Rott, P.; Warmuth, C.J.; Chatenet, M.; Baudin, P. Intraspecific genomic variation within Xanthomonas albilineans, the sugarcane leaf scald pathogen. Phytopathology 1997, 87, 316–324. [Google Scholar] [CrossRef]
- Pieretti, I.; Royer, M.; Barbe, V.; Carrere, S.; Koebnik, R.; Couloux, A.; Darrasse, A.; Gouzy, J.; Jacques, M.-A.; Lauber, E.; et al. Genomic insights into strategies used by Xanthomonas albilineans with its reduced artillery to spread within sugarcane xylem vessels. BMC Genom. 2012, 13, 658. [Google Scholar] [CrossRef]
- Lin, L.-H.; Ntambo, M.S.; Rott, P.C.; Wang, Q.-N.; Lin, Y.-H.; Fu, H.-Y.; Gao, S.-J. Molecular detection and prevalence of Xanthomonas albilineans, the causal agent of sugarcane leaf scald, in China. Crop Prot. 2018, 109, 17–23. [Google Scholar] [CrossRef]
- Ntambo, M.S.; Meng, J.-Y.; Rott, P.C.; Royer, M.; Lin, L.-H.; Zhang, H.-L.; Gao, S.-J. Identification and characterization of Xanthomonas albilineans causing sugarcane leaf scald in China using multilocus sequence analysis. Plant Pathol. 2019, 68, 269–277. [Google Scholar] [CrossRef]
- Bansal, K.; Midha, S.; Kumar, S.; Patil, P.B. Ecological and evolutionary insights into Xanthomonas citri pathovar diversity. Appl. Environ. Microbiol. 2017, 83, e02993-16. [Google Scholar] [CrossRef] [PubMed]
- Pieretti, I.; Royer, M.; Barbe, V.; Carrere, S.; Koebnik, R.; Cociancich, S.; Couloux, A.; Darrasse, A.; Gouzy, J.; Jacques, M.-A.; et al. The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. Bmc Genom. 2009, 10, 616. [Google Scholar] [CrossRef] [PubMed]
- Giampetruzzi, A.; Saponari, M.; Loconsole, G.; Boscia, D.; Savino, V.N.; Almeida, R.P.P.; Zicca, S.; Landa, B.B.; Chacon-Diaz, C.; Saldarelli, P. Genome-wide analysis provides evidence on the genetic relatedness of the emergent Xylella fastidiosa genotype in Italy to isolates from central America. Phytopathology 2017, 107, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Shan, H.L.; Li, W.F.; Cang, X.Y.; Wang, X.Y.; Yin, J.; Luo, Z.M.; Huang, Y.K. First report of sugarcane leaf scald caused by Xanthomonas albilineans (Ashby) Dowson in the province of Guangxi, China. Plant Dis. 2017, 101. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Wang, X.-Y.; Shan, H.-L.; Li, J.; Li, W.-F.; Cang, X.-Y.; Luo, Z.-M.; Yin, J.; Huang, Y.-K. Identification and phylogenetic analysis of Xanthomonas albilineans (Ashby) Dowson based on multiple gene sequences in Yunnan province, China. Sugar Tech 2019, 21, 794–801. [Google Scholar] [CrossRef]
- Davis, M.J.; Rott, P.; Baudin, P.; Dean, J. Evaluation of selective media and immunoassays for detection of Xanthomonas albilineans, causal agent of sugarcane leaf scald disease. Plant Dis. 1994, 78, 78–82. [Google Scholar] [CrossRef]
- Lim, H.J.; Lee, E.H.; Yoon, Y.; Chua, B.; Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Kolbe, D.L.; Eddy, S.R. Infernal 1.0: Inference of RNA alignments. Bioinformatics 2009, 25, 1335–1337. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Saier, M.H., Jr.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, D645–D655. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Pop, M. ARDB--Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Ge, R.; Mai, G.; Wang, P.; Zhou, M.; Luo, Y.; Cai, Y.; Zhou, F. CRISPRdigger: Detecting CRISPRs with better direct repeat annotations. Sci. Rep. 2016, 6, 32942. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Richter, M.; Rossello-Mora, R.; Oliver Glockner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Chiaromonte, F.; Yap, V.; Miller, W. Scoring pairwise genomic sequence alignments. In Biocomputing 2002; World Scientific: Singapore, 2001; pp. 115–126. [Google Scholar]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef]
- Ferrarini, M.; Moretto, M.; Ward, J.A.; Surbanovski, N.; Stevanovic, V.; Giongo, L.; Viola, R.; Cavalieri, D.; Velasco, R.; Cestaro, A.; et al. An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genom. 2013, 14, 670. [Google Scholar] [CrossRef]
- Rott, P.; Fleites, L.; Marlow, G.; Royer, M.; Gabriel, D.W. Identification of new candidate pathogenicity factors in the xylem-invading pathogen Xanthomonas albilineans by transposon mutagenesis. Mol. Plant Microbe Interact. 2011, 24, 594–605. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, W.; Zhang, Y.; Wang, S.; Ma, R.; Sun, W. Ax21-triggered immunity plays a significant role in rice defense against Xanthomonas oryzae pv. oryzicola. Phytopathology 2013. [Google Scholar] [CrossRef]
- Bahar, O.; Pruitt, R.; Luu, D.D.; Schwessinger, B.; Daudi, A.; Liu, F.; Ruan, R.; Fontaine-Bodin, L.; Koebnik, R.; Ronald, P. The Xanthomonas Ax21 protein is processed by the general secretory system and is secreted in association with outer membrane vesicles. PeerJ 2014, 2, e242. [Google Scholar] [CrossRef]
- Shen, Y.; Sharma, P.; da Silva, F.G.; Ronald, P. The Xanthomonas oryzae pv. oryzae raxP and raxQ genes encode an ATP sulphurylase and adenosine-5′-phosphosulphate kinase that are required for AvrXa21 avirulence activity. Mol. Microbiol. 2002, 44, 37–48. [Google Scholar] [CrossRef]
- Bobay, L.M.; Ochman, H. The evolution of bacterial genome architecture. Front. Genet. 2017, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Mahony, J.; Ainsworth, S.; Nauta, A.; van Sinderen, D. Bacteriophage orphan DNA methyltransferases: Insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 2013, 79, 7547–7555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chakrabarty, P.K.; Fleites, L.A.; Rayside, P.A.; Hopkins, D.L.; Gabriel, D.W. Three new Pierce’s disease pathogenicity effectors identified using Xylella fastidiosa biocontrol strain EB92-1. Plos ONE 2015, 10, e0133796. [Google Scholar] [CrossRef]
- Pieretti, I.; Pesic, A.; Petras, D.; Royer, M.; Sussmuth, R.D.; Cociancich, S. What makes Xanthomonas albilineans unique amongst xanthomonads? Front. Plant Sci. 2015, 6, 289. [Google Scholar] [CrossRef]
- Bullman, S.; Lucid, A.; Corcoran, D.; Sleator, R.D.; Lucey, B. Genomic investigation into strain heterogeneity and pathogenic potential of the emerging gastrointestinal pathogen Campylobacter ureolyticus. Plos ONE 2013, 8, e71515. [Google Scholar] [CrossRef]
- Jangam, D.; Feschotte, C.; Betran, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Martins, P.M.; Machado, M.A.; Silva, N.V.; Takita, M.A.; de Souza, A.A. Type II toxin-antitoxin distribution and adaptive aspects on Xanthomonas genomes: Focus on Xanthomonas citri. Front. Microbiol. 2016, 7, 652. [Google Scholar] [CrossRef]
- Xiao, J.; Worby, C.A.; Mattoo, S.; Sankaran, B.; Dixon, J.E. Structural basis of Fic-mediated adenylylation. Nat. Struct. Mol. Biol. 2010, 17, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Mattoo, S.; Kruger, R.P.; Corbeil, L.B.; Koller, A.; Mendez, J.C.; Zekarias, B.; Lazar, C.; Dixon, J.E. The fic domain: Regulation of cell signaling by adenylylation. Mol. Cell 2009, 34, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Scheurwater, E.; Reid, C.W.; Clarke, A.J. Lytic transglycosylases: Bacterial space-making autolysins. Int. J. Biochem. Cell Biol. 2008, 40, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Dik, D.A.; Marous, D.R.; Fisher, J.F.; Mobashery, S. Lytic transglycosylases: Concinnity in concision of the bacterial cell wall. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 503–542. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element and Characteristics | Xa-FJ1 | GPE PC73 |
|---|---|---|
| Sequencing platform | PacBio RSII, Illumina PE150 | Sanger |
| Coverage | 206×, 570× | 17× |
| Size (bp) | 3,724,581 | 3,768,695 |
| G + C content (%) | 63 | 63 |
| No. protein-coding sequences (CDSs) | 3176 | 3115 |
| Coding density (%) | 86.66 | 84 |
| Average length in bp of all CDSs | 1016 | 1059 |
| Region | Xa-FJ1 | GPE PC73 | Variation Type (Reference = Xa-FJ1) | Fragment Length (nt) | Predicted Cause of Variation | Affected Gene(s) | |||
|---|---|---|---|---|---|---|---|---|---|
| Begins | Ends | Begins | Ends | Xa-FJ1 | GPE PC73 | ||||
| R1 | 219,400 | 219,400 | 219,691 | 271,791 | Insertion | 0 | 52,101 | Prophage integration | XALc_0171-XALc_0242 (72 specific genes in GPE PC73) |
| R2 | 679,518 | 681,202 | 731,933 | 731,933 | Deletion | 1685 | 0 | Recombination/assembly defect | XaFJ1_GM000644; XaFJ1_GM000645; XaFJ1_GM000646 |
| R3 | 1,165,743 | 1,168,928 | 1,216,465 | 1,216,465 | Deletion | 3186 | 0 | Recombination/assembly defect | XaFJ1_GM001035; XALc_1056 |
| R4 | 1,727,177 | 1,727,177 | 1,774,582 | 1,780,815 | Insertion | 0 | 6234 | Recombination/assembly defect | XaFJ1_GM001510; XALc_1529 |
| R5 | 1,736,246 | 1,736,755 | 1,789,885 | 1,790,986 | Complex Indel | 510 | 1102 | Prophage integration | XALC_1536-XALC_1545 (10 specific genes in GPE PC73); XaFJ1_GM001517-XaFJ1_GM001525 (9 specific genes in Xa-FJ1) |
| R6 | 1,736,873 | 1,741,929 | 1,791,101 | 1,795,842 | Complex Indel | 5057 | 4742 | Prophage integration | |
| R7 | 1,742,065 | 1,742,065 | 1,795,979 | 1,796,122 | Insertion | 0 | 144 | Prophage integration | |
| R8 | 1,742,742 | 1,742,742 | 1,796,839 | 1,797,116 | Insertion | 0 | 278 | Prophage integration | |
| R9 | 1,778,246 | 1,784,662 | 1,832,620 | 1,832,620 | Deletion | 6417 | 0 | Recombination/assembly defect | XaFJ1_GM001532; XALc_1551 |
| R10 | 2,484,556 | 2,484,556 | 2,532,506 | 2,535,350 | Insertion | 0 | 2845 | Recombination/assembly defect | XaFJ1_GM002152; XALc_2151; XALc_2152 |
| R11 | 26,75,326 | 2,679,992 | 2,726,120 | 2,726,120 | Deletion | 4667 | 0 | Transposable elements | XaFJ1_GM002293; XaFJ1_GM002292; XaFJ1_GM002291; XALc_2290 |
| R12 | 3,041,680 | 3,041,680 | 3,087,815 | 3,088,684 | Insertion | 0 | 870 | Transposable elements | XALc_2603; XALc_2604 |
| R13 | 3,377,378 | 3,377,378 | 3,424,367 | 3,424,696 | Insertion | 0 | 330 | CRISPR-Cas | Intergenic region which contains clustered regulatory interspaced short palindromic repeats |
| R14 | 3,377,647 | 3,377,713 | 3,424,965 | 3,424,965 | Deletion | 67 | 0 | CRISPR-Cas | |
| R15 | 3,377,912 | 3,377,912 | 3,425,163 | 3,425,227 | Insertion | 0 | 65 | CRISPR-Cas | |
| R16 | 3,460,518 | 3,463,513 | 3,507,852 | 3,507,852 | Deletion | 2996 | 0 | Transposable elements | XaFJ1_GM002988; XaFJ1_GM002989; XaFJ1_GM002990; XALc_2969 |
| Specific Gene | Annotation | Location |
|---|---|---|
| XaFJ1_GM001517 | zona occludens toxin | the plasticity zone, R5-R8 |
| XaFJ1_GM001518 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001519 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001520 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001521 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001522 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001523 | DNA-binding protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001524 | replication initiation protein | the plasticity zone, R5-R8 |
| XaFJ1_GM001525 | hypothetical protein | the plasticity zone, R5-R8 |
| XaFJ1_GM002989 | transposase | transposable elements, R16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-L.; Ntambo, M.S.; Rott, P.C.; Chen, G.; Chen, L.-L.; Huang, M.-T.; Gao, S.-J. Complete Genome Sequence Reveals Evolutionary and Comparative Genomic Features of Xanthomonas albilineans Causing Sugarcane Leaf Scald. Microorganisms 2020, 8, 182. https://doi.org/10.3390/microorganisms8020182
Zhang H-L, Ntambo MS, Rott PC, Chen G, Chen L-L, Huang M-T, Gao S-J. Complete Genome Sequence Reveals Evolutionary and Comparative Genomic Features of Xanthomonas albilineans Causing Sugarcane Leaf Scald. Microorganisms. 2020; 8(2):182. https://doi.org/10.3390/microorganisms8020182
Chicago/Turabian StyleZhang, Hui-Li, Mbuya Sylvain Ntambo, Philippe C. Rott, Gongyou Chen, Li-Lan Chen, Mei-Ting Huang, and San-Ji Gao. 2020. "Complete Genome Sequence Reveals Evolutionary and Comparative Genomic Features of Xanthomonas albilineans Causing Sugarcane Leaf Scald" Microorganisms 8, no. 2: 182. https://doi.org/10.3390/microorganisms8020182
APA StyleZhang, H.-L., Ntambo, M. S., Rott, P. C., Chen, G., Chen, L.-L., Huang, M.-T., & Gao, S.-J. (2020). Complete Genome Sequence Reveals Evolutionary and Comparative Genomic Features of Xanthomonas albilineans Causing Sugarcane Leaf Scald. Microorganisms, 8(2), 182. https://doi.org/10.3390/microorganisms8020182