Salmonella Genomic Island 1 is Broadly Disseminated within Gammaproteobacteriaceae




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Preliminary Screening of SRA for SGI1-REs and Subsequent Genome Assembly
2.2. Putative Identification and Clustering of SGI1-RE Variants
2.3. Annotation and Comparative Analysis of SGI1-REs
2.4. Phylogenomic Analysis and Genotypic Characterization of Clonal Subpopulations
2.5. Data Availability
3. Results and Discussion
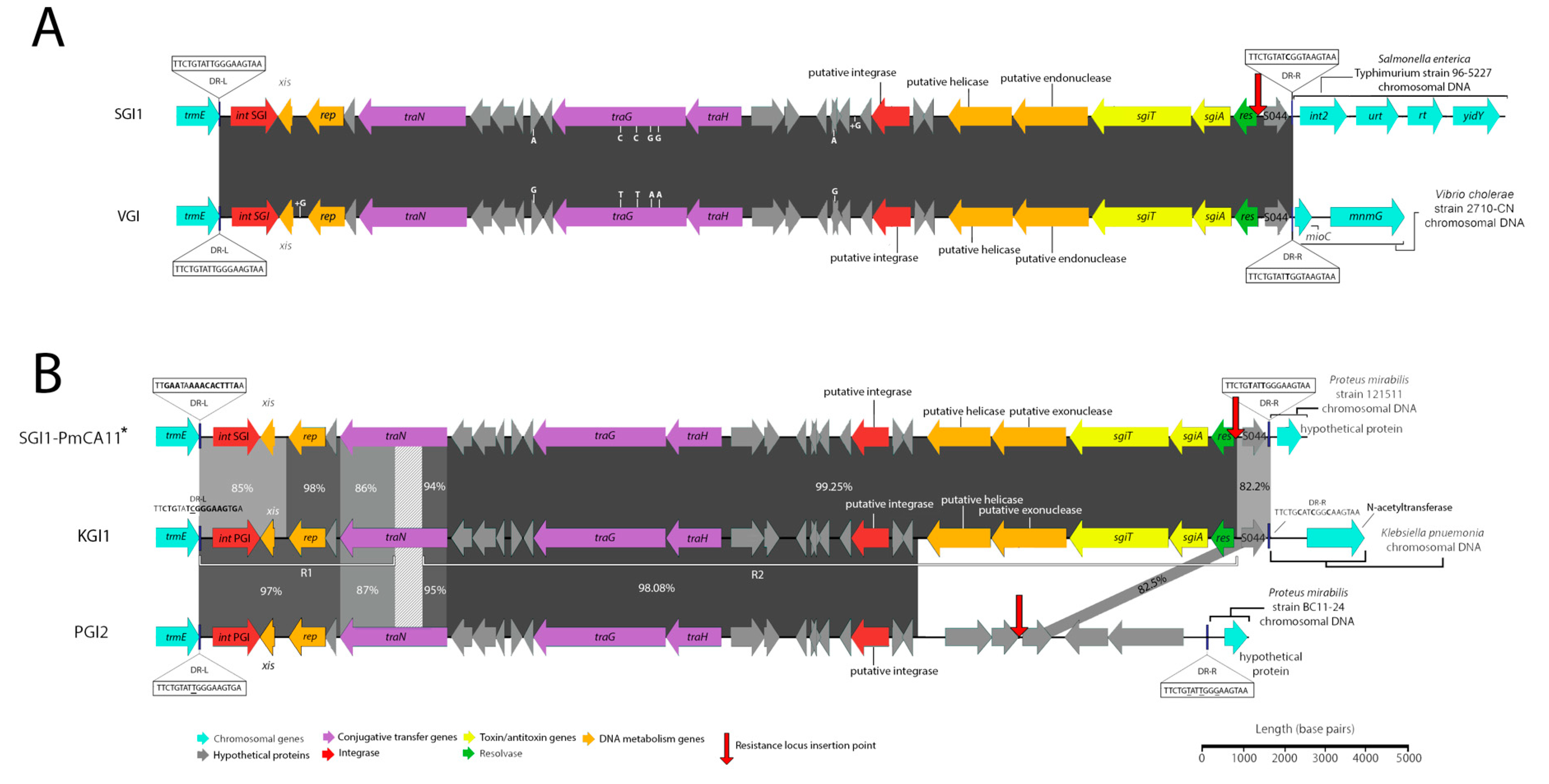
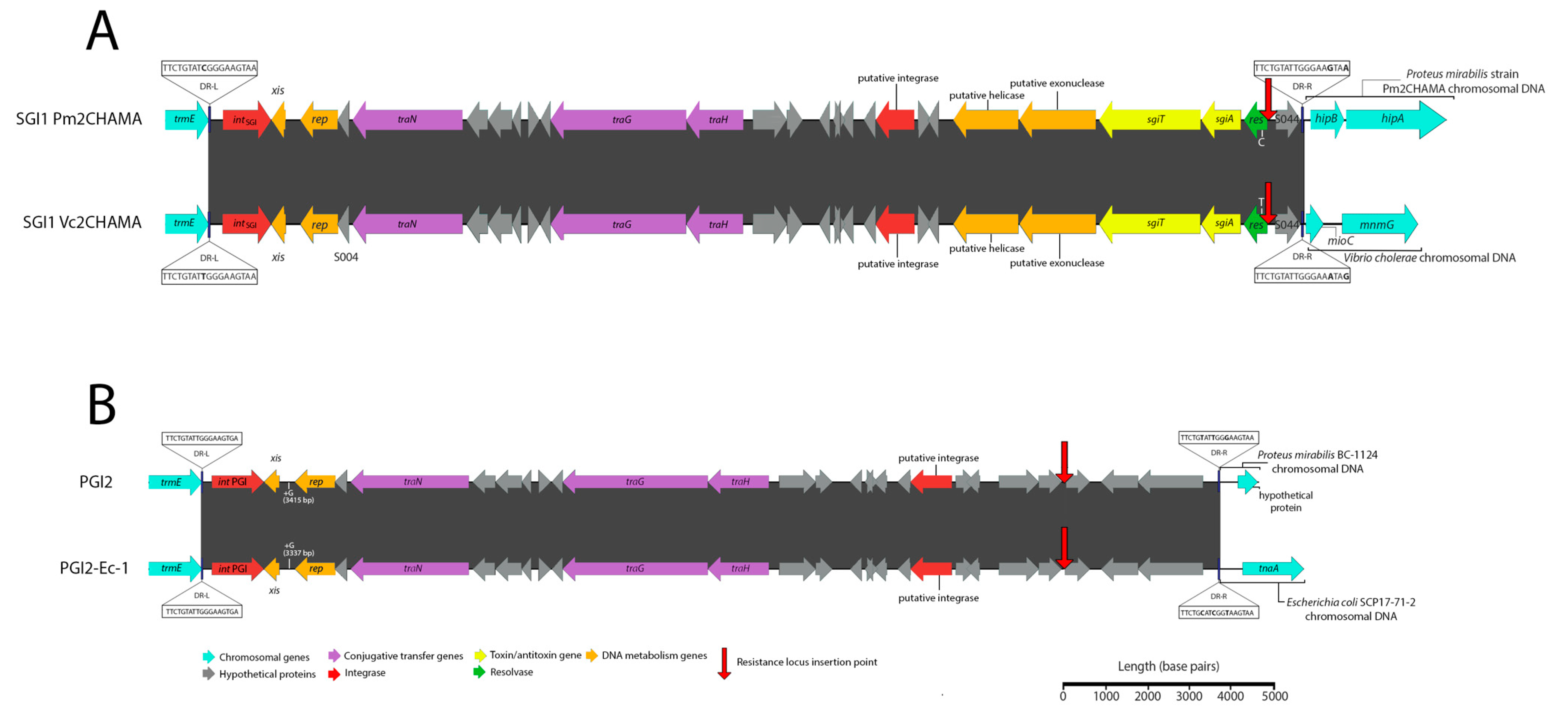
3.1. Potential Discovery of an Ancestral Variant of Salmonella Genomic Island 1
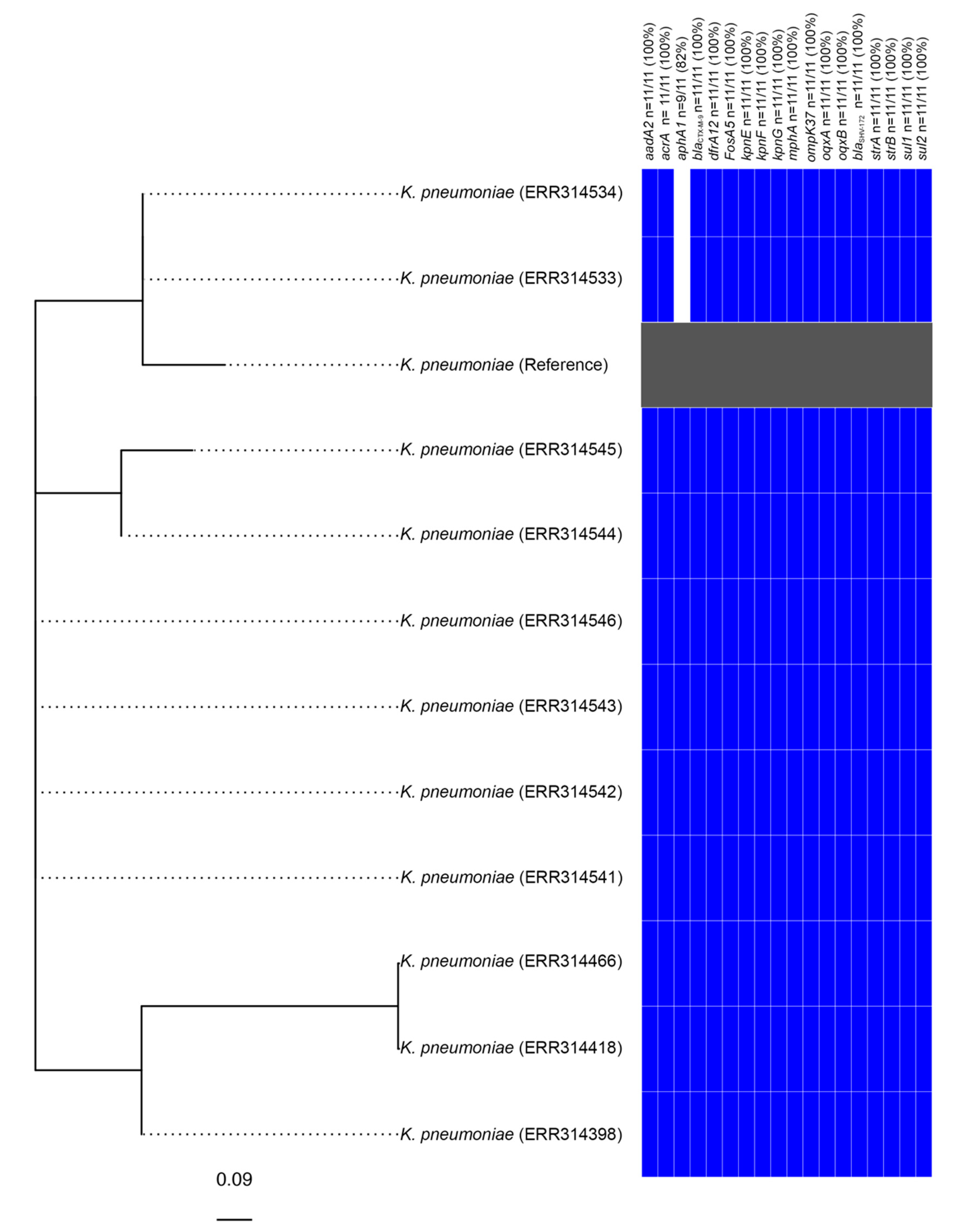
3.2. Identification of a Clonal Outbreak of Extensively Drug-Resistant Klebsiella pneumoniae Carrying a Novel SGI1-RE
3.3. Novel Variants and Hosts of SGI1-Related Elements
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malcova, M.; Hradecka, H.; Karpiskova, R.; Rychlik, I. Biofilm formation in field strains of Salmonella enterica serovar Typhimurium: Identification of a new colony morphology type and the role of SGI1 in biofilm formation. Vet. Microbiol. 2008, 129, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Lei, C.W.; Kang, Z.Z.; Zhang, Y.; Wang, H.-N. IS26-mediated genetic rearrangements in Salmonella genomic island 1 of Proteus mirabilis. Front. Microbiol. 2019, 10, 2245. [Google Scholar] [CrossRef] [PubMed]
- Hamidian, M.; Holt, K.E.; Hall, R.M. Genomic resistance island AGI1 carrying a complex class 1 integron in a multiply antibiotic-resistant ST25 Acinetobacter baumannii isolate. J. Antimicrob. Chemother. 2015, 70, 2519–2523. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.M.; Shimamoto, T.; Nariya, H.; Shimamoto, T. Emergence of Salmonella genomic island 1 variant SGI1-W in a clinical isolate of Providencia stuartii from Egypt. Antimicrob. Agents Chemother. 2018. [Google Scholar] [CrossRef]
- Siebor, E.; de Curraize, C.; Neuwirth, C. Identification of AGI1-A, a variant of Acinetobacter genomic island 1 (AGI1), in a French clinical isolate belonging to the Enterobacter cloacae complex. J. Antimicrob. Chemother. 2018, 74, 311–314. [Google Scholar] [CrossRef]
- Cummins, M.L.; Chowdhury, P.R.; Marenda, M.S.; Browning, G.F.; Djordjevic, S.P. Salmonella Genomic Island 1B Variant Found in a Sequence Type 117 Avian Pathogenic Escherichia coli Isolate. mSphere 2019, 4, e00169-19. [Google Scholar] [CrossRef]
- Siebor, E.; de Curraize, C.; Amoureux, L.; Neuwirth, C. Mobilization of the Salmonella genomic island SGI1 and the Proteus genomic island PGI1 by the A/C2 plasmid carrying blaTEM-24 harboured by various clinical species of Enterobacteriaceae. J. Antimicrob. Chemother. 2016, 71, 2167–2170. [Google Scholar] [CrossRef]
- Doublet, B.; Golding, G.R.; Mulvey, M.R.; Cloeckaert, A. Potential integration sites of the Salmonella genomic island 1 in Proteus mirabilis and other bacteria. J. Antimicrob. Chemother. 2007, 59, 801–803. [Google Scholar] [CrossRef]
- Bradley, P.; Den Bakker, H.; Rocha, E.; McVean, G.; Iqbal, Z. Real-time search of all bacterial and viral genomic data. BioRxiv 2017. BioRxiv:234955. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- Cummins, M.L.; Reid, C.J.; Chowdhury, P.R.; Bushell, R.N.; Esbert, N.; Tivendale, K.A.; Noormohammadi, A.H.; Islam, S.; Marenda, M.S.; Browning, G.F.; et al. Whole genome sequence analysis of Australian avian pathogenic Escherichia coli that carry the class 1 integrase gene. Microb. Genom. 2019, 5, e000250. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- de Curraize, C.; Siebor, E.; Neuwirth, C.; Hall, R.M. SGI0, a relative of Salmonella genomic islands SGI1 and SGI2, lacking a class 1 integron, found in Proteus mirabilis. Plasmid 2020, 107, 102453. [Google Scholar] [CrossRef]
- Xiao, T.; Dai, H.; Lu, B.; Li, Z.; Cai, H.; Huang, Z.; Kan, B.; Wang, D. Distribution and characteristics of SGI1/PGI2 genomic island from Proteus strains in China. Infect. Genet. Evol. 2019, 70, 123–130. [Google Scholar] [CrossRef]
- Ellington, M.J.; Heinz, E.; Wailan, A.M.; Dorman, M.J.; De Goffau, M.; Cain, A.K.; Henson, S.P.; Gleadall, N.; Boinett, C.J.; Dougan, G.; et al. Contrasting patterns of longitudinal population dynamics and antimicrobial resistance mechanisms in two priority bacterial pathogens over 7 years in a single center. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Kluytmans-van den Bergh, M.F.; Rossen, J.W.; Bruijning-Verhagen, P.C.; Bonten, M.J.; Friedrich, A.W.; Vandenbroucke-Grauls, C.M.J.E.; Willems, R.J.L.; Kluytmans, J.A.J.W.; the SoM Study Group. Whole-genome multilocus sequence typing of extended-spectrum-beta-lactamase-producing Enterobacteriaceae. J. Clin. Microbiol. 2016, 54, 2919–2927. [Google Scholar] [CrossRef]
- Partridge, S.R.; Brown, H.J.; Stokes, H.W.; Hall, R.M. Transposons Tn1696 and Tn21 and their integrons In4 and In2 have independent origins. Antimicrob. Agents Chemother. 2001, 45, 1263–1270. [Google Scholar] [CrossRef]
- Partridge, S.R.; Recchia, G.D.; Stokes, H.W.; Hall, R.M. Family of class 1 integrons related to In4 from Tn1696. Antimicrob. Agents Chemother. 2001, 45, 3014–3020. [Google Scholar] [CrossRef]
- Qiu, D.; Wei, H.; Tu, Q.; Yang, Y.; Xie, M.; Chen, J.; Pinkerton, M.H., Jr.; Liang, Y.; He, Z.; Zhou, J. Combined genomics and experimental analyses of respiratory characteristics of Shewanella putrefaciens W3-18-1. Appl. Environ. Microbiol. 2013, 79, 5250–5257. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cummins, M.L.; Hamidian, M.; Djordjevic, S.P. Salmonella Genomic Island 1 is Broadly Disseminated within Gammaproteobacteriaceae. Microorganisms 2020, 8, 161. https://doi.org/10.3390/microorganisms8020161
Cummins ML, Hamidian M, Djordjevic SP. Salmonella Genomic Island 1 is Broadly Disseminated within Gammaproteobacteriaceae. Microorganisms. 2020; 8(2):161. https://doi.org/10.3390/microorganisms8020161
Chicago/Turabian StyleCummins, Max Laurence, Mohammad Hamidian, and Steven Philip Djordjevic. 2020. "Salmonella Genomic Island 1 is Broadly Disseminated within Gammaproteobacteriaceae" Microorganisms 8, no. 2: 161. https://doi.org/10.3390/microorganisms8020161
APA StyleCummins, M. L., Hamidian, M., & Djordjevic, S. P. (2020). Salmonella Genomic Island 1 is Broadly Disseminated within Gammaproteobacteriaceae. Microorganisms, 8(2), 161. https://doi.org/10.3390/microorganisms8020161