Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea

 ,
, 
 , and
, and 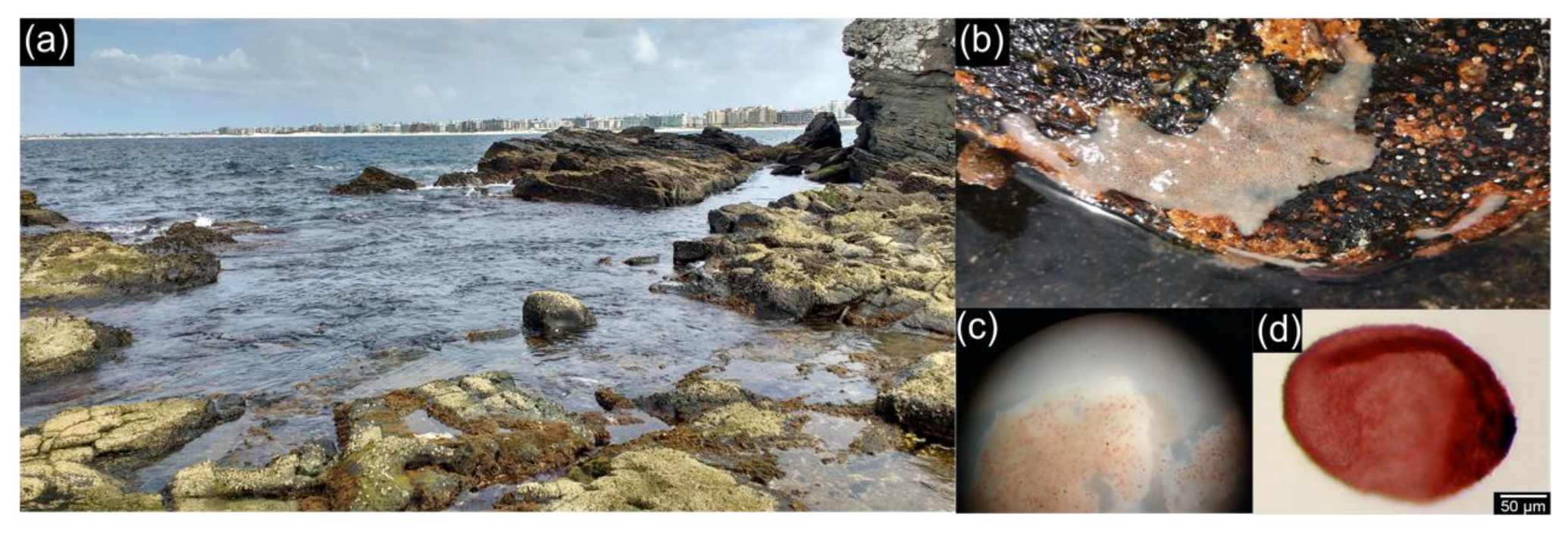{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection and Processing of the Samples
2.2. DNA Extraction and Sequencing
2.3. Bioinformatic Analyses
2.4. Statistical Analyses
2.5. Analyses of Sponge-Enriched OTUs
2.6. Global Analyses of Homoscleromorpha-Associated Prokaryotic Microbiota
2.7. Molecular Sponge Identification
3. Results
3.1. Molecular Sponge Taxonomy
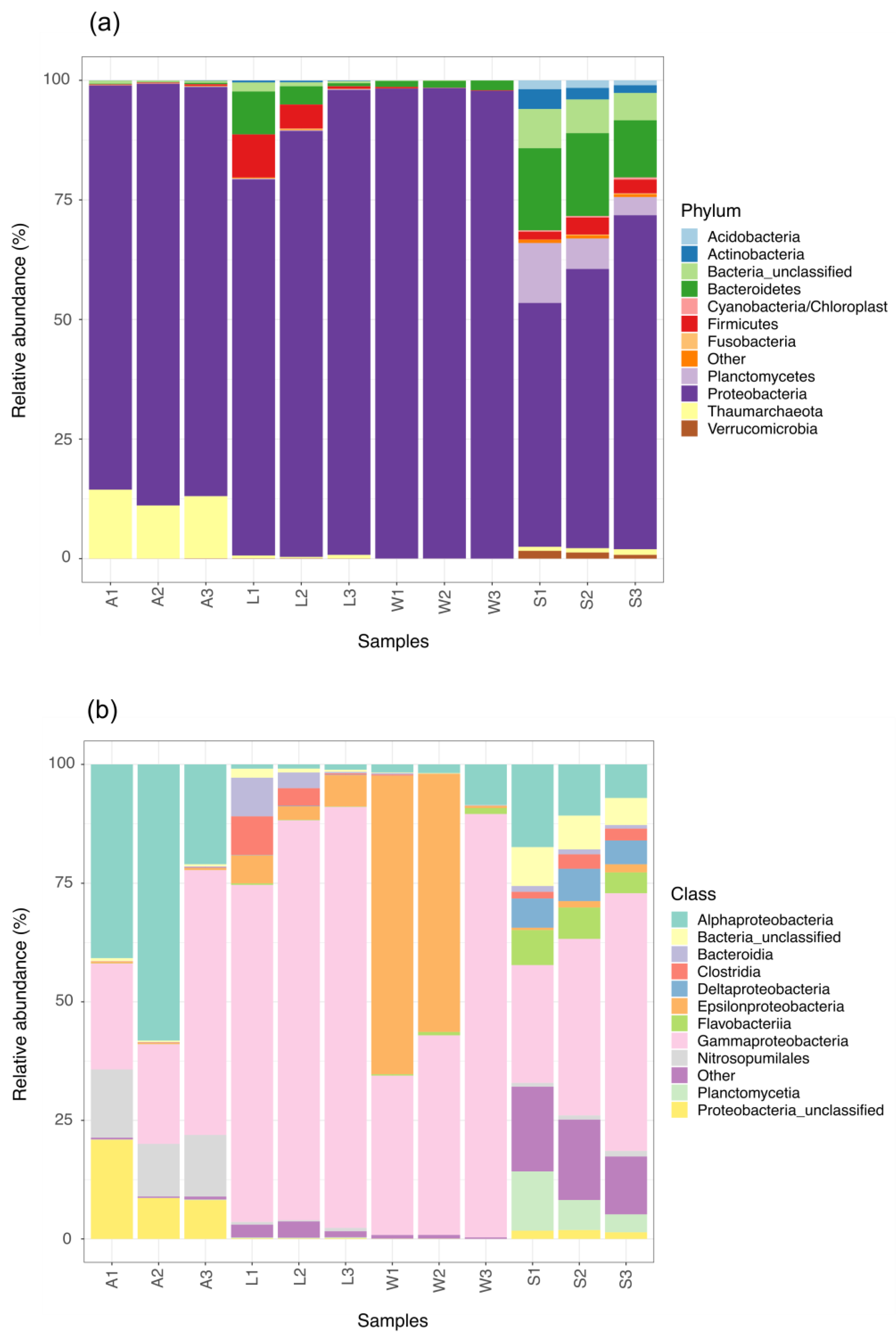
3.2. Prokaryotic Community Composition
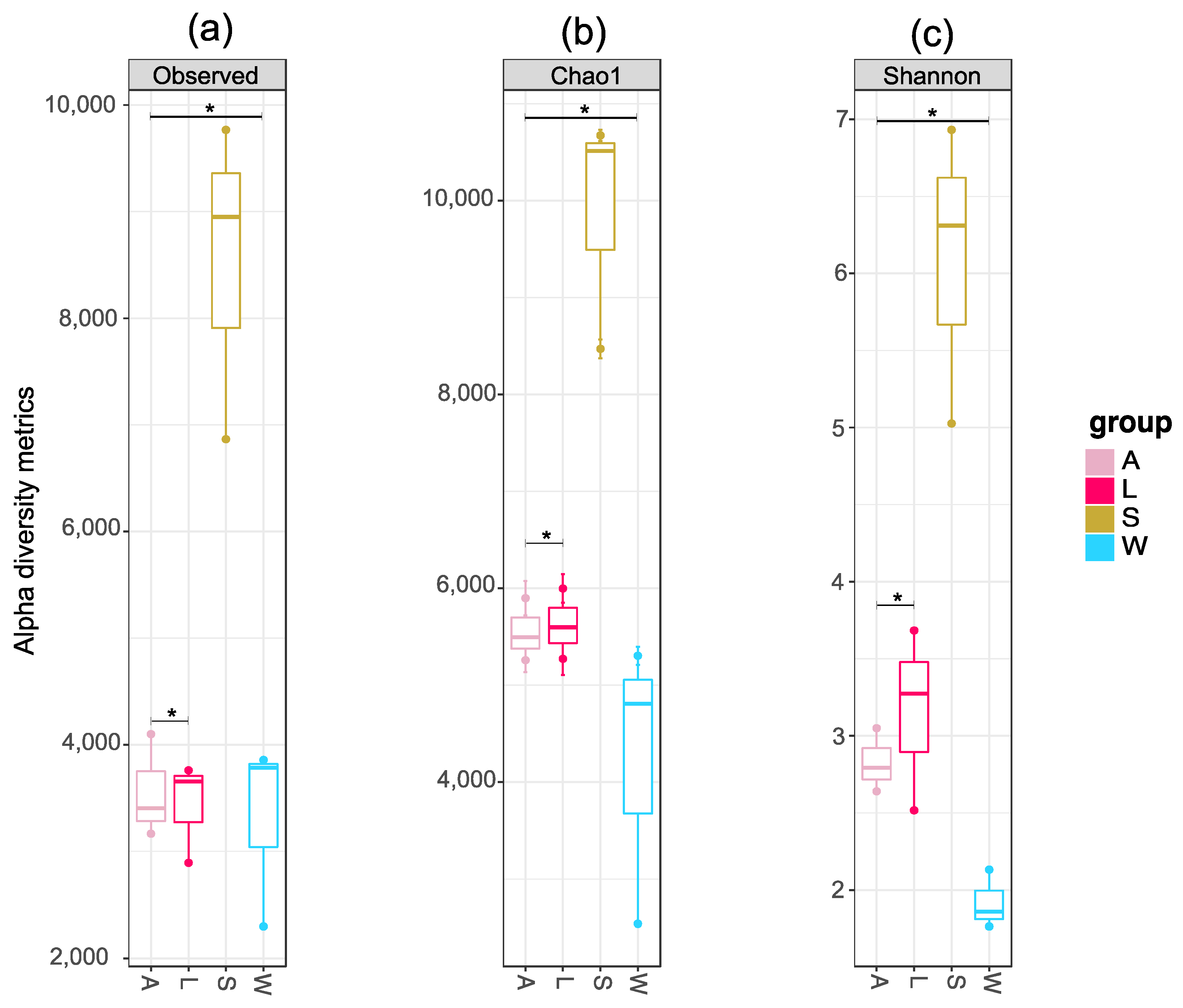
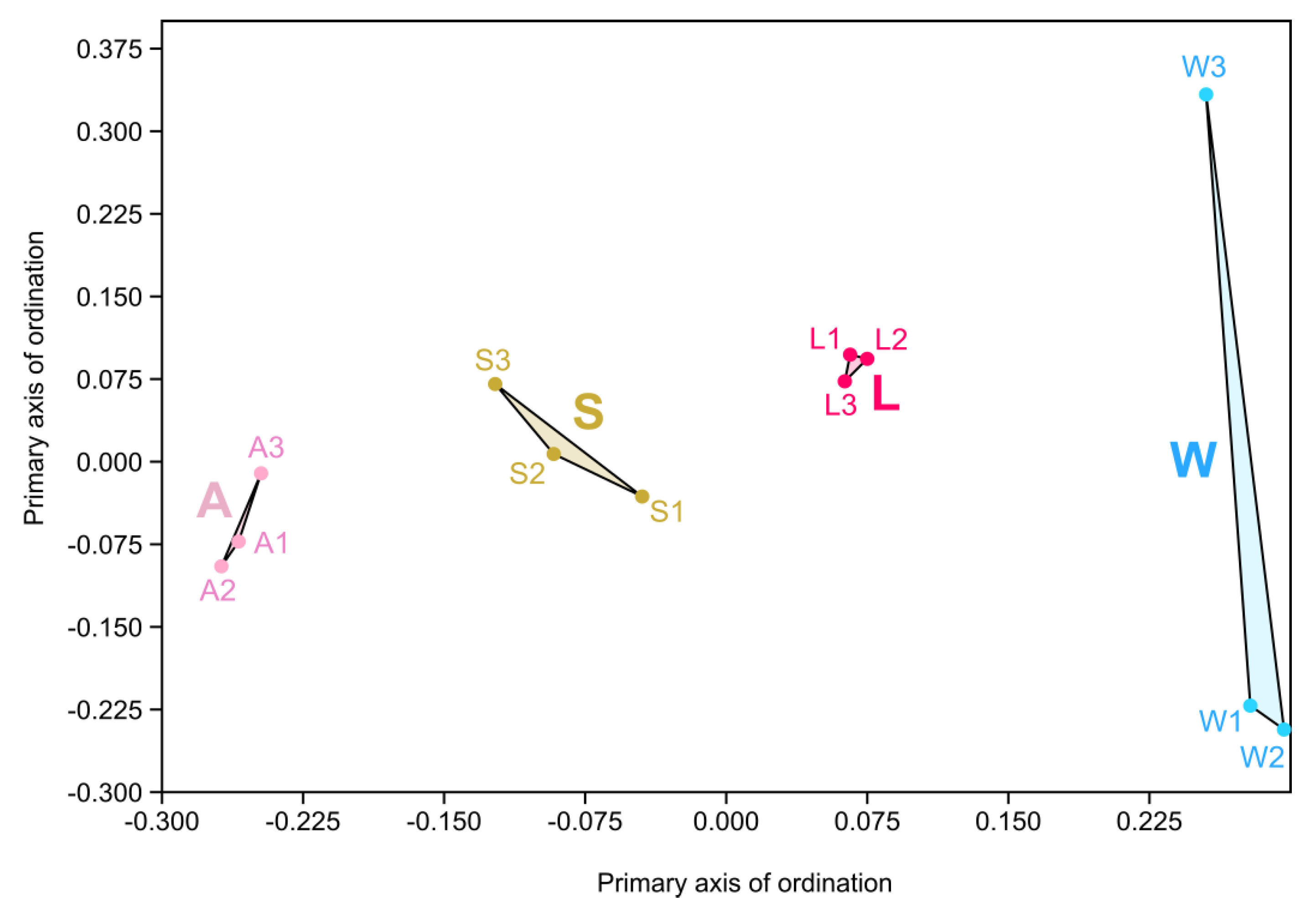
3.3. Prokaryotic Community Diversity and Structure
3.4. OTU-Level and Core Microbiota Analyses
3.5. Global Analyses of Homoscleromorpha-Associated Prokaryotic Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Richardson, L.A. Evolving as a holobiont. PLoS Biol. 2017, 15, e2002168. [Google Scholar] [CrossRef]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.; Zilber-Rosenberg, I. The hologenome concept of evolution after 10 years. Microbiome 2018, 6, 78. [Google Scholar] [CrossRef] [PubMed]
- Bright, M.; Bulgheresi, S. A complex journey: Transmission of microbial symbionts. Nat. Rev. Microbiol. 2010, 8, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.C.; Marchesi, J.R.; Mougel, C.; Selosse, M.A. Host-microbiota interactions: From holobiont theory to analysis. Microbiome 2019, 7, 5. [Google Scholar] [CrossRef]
- Vrijenhoek, R.C. Genetics and evolution of deep-sea chemosynthetic bacteria and their invertebrate hosts. In The Vent and Seep Biota, 1st ed.; Steffen, K., Ed.; Springer: Dordrecht, Germany, 2010; Volume 33, pp. 15–49. [Google Scholar] [CrossRef]
- Sachs, J.L.; Skophammer, R.G.; Regus, J.U. Evolutionary transitions in bacterial symbiosis. Proc. Natl. Acad. Sci. USA 2011, 108, 10800–10807. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E.; Werren, J.H. Holes in the hologenome: Why host-microbe symbioses are not holobionts. mBio 2016, 7, e02099-15. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D. The epidemiology and evolution of symbionts with mixed-mode transmission. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 623–643. [Google Scholar] [CrossRef]
- Haag, K.L. Holobionts and their hologenomes: Evolution with mixed modes of inheritance. Genet. Mol. 2018, 41. [Google Scholar] [CrossRef]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-associated microorganisms: Evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef]
- Pita, L.; Rix, L.; Slaby, B.M.; Franke, A.; Hentschel, U. The sponge holobiont in a changing ocean: From microbes to ecosystems. Microbiome 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Jonas, L.; Lin, H.; Hill, R.T. Microbially mediated nutrient cycles in marine sponges. FEMS Microbiol. Ecol. 2019, 95, fiz155. [Google Scholar] [CrossRef] [PubMed]
- Thacker, R.W.; Freeman, C.J. Sponge–microbe symbioses: Recent advances and new directions. In Advances in Marine Biology, 1st ed.; Lesser, M., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 62, pp. 57–111. [Google Scholar] [CrossRef]
- Webster, N.S.; Thomas, T. The sponge hologenome. mBio 2016, 7. [Google Scholar] [CrossRef]
- Webster, N.S.; Taylor, M.W.; Behnam, F.; Lücker, S.; Rattei, T.; Whalan, S.; Wagner, M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ. Microbiol. 2010, 12, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Turon, M.; Cáliz, J.; Garate, L.; Casamayor, E.O.; Uriz, M.J. Showcasing the role of seawater in bacteria recruitment and microbiome stability in sponges. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Wu, S.; Ou, H.; Liu, T.; Wang, D.; Zhao, J. Structure and dynamics of microbiomes associated with the marine sponge Tedania sp. during its life cycle. FEMS Microbiol. Ecol. 2018, 94, fiy055. [Google Scholar] [CrossRef]
- Sacristán-Soriano, O.; Winkler, M.; Erwin, P.; Weisz, J.; Harriott, O.; Heussler, G.; Hill, M. Ontogeny of symbiont community structure in two carotenoid-rich, viviparous marine sponges: Comparison of microbiomes and analysis of culturable pigmented heterotrophic bacteria. Environ. Microbial. Rep. 2019, 11, 249–261. [Google Scholar] [CrossRef]
- Björk, J.R.; Diéz-Vives, C.; Astudillo-García, C.; Archie, E.A.; Montoya, J.M. Vertical transmission of sponge microbiota is inconsistent and unfaithful. Nat. Ecol. Evol. 2019, 3, 1172–1183. [Google Scholar] [CrossRef]
- Hentschel, U.; Hopke, J.; Horn, M.; Friedrich, A.B.; Wagner, M.; Hacker, J.; Moore, B.S. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl. Environ. Microbial. 2002, 68, 4431–4440. [Google Scholar] [CrossRef]
- Taylor, M.W.; Tsai, P.; Simister, R.L.; Deines, P.; Botte, E.; Ericson, G.; Webster, N.S. ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 2013, 7, 438–443. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Bayer, K.; Cannistraci, C.V.; Giles, E.C.; Ryu, T.; Seridi, L.; Hentschel, U. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 2014, 23, 1348–1363. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Angermeier, H.; Schiller, R.; Lindquist, N.; Hentschel, U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 2008, 74, 7694–7708. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Tsai, P.; Bell, J.; Fromont, J.; Ilan, M.; Lindquist, N.; Webster, N. Assessing the complex sponge microbiota: Core, variable and species-specific bacterial communities in marine sponges. ISME J. 2012, 6, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Sipkema, D.; de Caralt, S.; Morillo, J.A.; Al-Soud, W.A.; Sørensen, S.J.; Smidt, H.; Uriz, M.J. Similar sponge-associated bacteria can be acquired via both vertical and horizontal transmission. Environ. Microbiol. 2015, 17, 3807–3821. [Google Scholar] [CrossRef]
- Fieth, R.A.; Gauthier, M.E.A.; Bayes, J.; Green, K.M.; Degnan, S.M. Ontogenetic changes in the bacterial symbiont community of the tropical demosponge Amphimedon queenslandica: Metamorphosis is a new beginning. Front. Mar. Sci. 2016, 3, 228. [Google Scholar] [CrossRef]
- Gazave, E.; Lape’bie, P.; Renard, E.; Vacelet, J.; Rocher, C. Molecular Phylogeny Restores the Supra-Generic Subdivision of Homoscleromorph Sponges (Porifera, Homoscleromorpha). PLoS ONE 2010, 5, e14290. [Google Scholar] [CrossRef]
- Gazave, E.; Lapébie, P.; Ereskovsky, A.V.; Vacelet, J.; Renard, E.; Cárdenas, P.; Borchiellini, C. No longer Demospongiae: Homoscleromorph sponges revisited by molecular phylogeny, Linnaean classification and the PhyloCode. Hydrobiologia 2012, 687, 3–10. [Google Scholar] [CrossRef]
- Nielsen, C. Phylum Homoscleromorpha. In Animal Evolution: Interrelationships of the Living Phyla, 3rd ed.; Oxford University Press: Oxford, UK, 2012; Volume 53, pp. 8–9. [Google Scholar] [CrossRef]
- Renard, E.; Gazave, E.; Fierro-Constain, L.; Schenkelaars, Q.; Ereskovsky, A.; Vacelet, J.; Borchiellini, C. Porifera (sponges): Recent knowledge and new perspectives. eLS 2013. [Google Scholar] [CrossRef]
- Fidler, A.L.; Darris, C.E.; Chetyrkin, S.V.; Pedchenko, V.K.; Boudko, S.P.; Brown, K.L.; Hudson, B.G. Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. eLife 2017, 6, e24176. [Google Scholar] [CrossRef]
- Belahbib, H.; Renard, E.; Santini, S.; Jourda, C.; Claverie, J.M.; Borchiellini, C.; Le Bivic, A. New genomic data and analyses challenge the traditional vision of animal epithelium evolution. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Ereskovsky, A.V.; Lavrov, D.V.; Willenz, P. Five new species of Homoscleromorpha (Porifera) from the Caribbean Sea and re-description of Plakina Jamaicensis. J. Mar. 2014, 94, 285. [Google Scholar] [CrossRef]
- Boury-Esnault, N.; Lavrov, D.V.; Ruiz, C.A.; Pérez, T. The integrative taxonomic approach applied to Porifera: A case study of the Homoscleromorpha. Integr. Comp. Biol. 2013, 53, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Gerovasileiou, V.; Voultsiadou, E. Marine caves in the Mediterranean Sea (No. RefW-14-59632). AUTh 2013. [Google Scholar] [CrossRef]
- van Soest, R.W.M.; Boury-Esnault, N.; Hooper, J.N.A. World Porifera Database. Available online: http://www.marinespecies.org/porifera/porifera.php?p=stats (accessed on 25 November 2020).
- Ruiz, C.; Muricy, G.; Lage, A.; Domingos, C.; Chenesseau, S.; Pérez, T. Descriptions of new sponge species and genus, including aspiculate Plakinidae, overturn the Homoscleromorpha classification. Zool. J. Linnean Soc. 2017, 179, 707–724. [Google Scholar] [CrossRef]
- Grenier, M.; Ruiz, C.; Lage, A.; Perez, T. New cave-dwelling Plakina (Plakinidae, Homoscleromorpha, Porifera) from Martinique Island (French Antilles). Zootaxa 2020, 4729, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Lage, A.; Gerovasileiou, V.; Voultsiadou, E.; Muricy, G. Taxonomy of Plakina (Porifera: Homoscleromorpha) from Aegean submarine caves, with descriptions of three new species and new characters for the genus. Mar. Biodivers. 2018, 49, 727–747. [Google Scholar] [CrossRef]
- Ruiz, C.; Ivanišević, J.; Chevaldonné, P.; Ereskovsky, A.V.; Boury-Esnault, N.; Vacelet, J.; Thomas, O.P.; Pérez, T. Integrative taxonomic description of Plakina kanaky, a new polychromatic sponge species from New Caledonia (Porifera: Homoscleromorpha). Mar. Ecol. 2015, 36, 1129–1143. [Google Scholar] [CrossRef]
- Lage, A.; Muricy, G.; Ruiz, C.; Perez, T. New sciaphilic plakinids (Porifera, Homoscleromorpha) from the Central-Western Pacific. Zootaxa 2018, 4466, 8–38. [Google Scholar] [CrossRef]
- Belmonte, T.; Alvim, J.; Padula, V.; Muricy, G. Spongivory by nudibranchs on the coast of Rio de Janeiro state, southeastern Brazil. Spixiana 2015, 38, 187–195. [Google Scholar]
- Domingos, C.; Lage, A.; Muricy, G. Overview of the biodiversity and distribution of the Class Homoscleromorpha in the Tropical Western Atlantic. J. Mar. 2016, 96, 379. [Google Scholar] [CrossRef]
- Muricy, G.; Domingos, C.; Lage, A.; Lanna, E.; Hardoim, C.C.; Laport, M.S.; Zilberberg, C. Integrative taxonomy widens our knowledge of the diversity, distribution and biology of the genus Plakina (Homosclerophorida: Plakinidae). Invertebr. Syst. 2019, 33, 367–401. [Google Scholar] [CrossRef]
- Laport, M.S.; Bauwens, M.; de Oliveira Nunes, S.; Willenz, P.; George, I.; Muricy, G. Culturable bacterial communities associated to Brazilian Oscarella species (Porifera: Homoscleromorpha) and their antagonistic interactions. Antonie van Leeuwenhoek 2017, 110, 489–499. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.F.R.; Cavalcanti, M.D.A.; de Oliveira Nunes, S.; Lobo, L.A.; Domingues, R.M.C.P.; Muricy, G.; Laport, M.S. Paraclostridium is the main genus of anaerobic bacteria isolated from new species of the marine sponge Plakina in the Brazilian Southeast coast. Curr. Microbiol. 2019, 76, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Freitas-Silva, J.; Silva-Oliveira, T.; Muricy, G.; Laport, M.S. Bacillus strains associated to homoscleromorpha sponges are highly active against multidrug resistant bacteria. Curr. Microbiol. 2020, 1–9. [Google Scholar] [CrossRef]
- Ruiz, C.; Villegas-Plazas, M.; Thomas, O.P.; Junca, H.; Pérez, T. Specialized microbiome of the cave-dwelling sponge Plakina kanaky (Porifera, Homoscleromorpha). FEMS Microbiol. Ecol. 2020, 96, fiaa043. [Google Scholar] [CrossRef]
- Fukami, H.; Budd, A.F.; Levitan, D.R.; Jara, J.; Kersanach, R.; Knowlton, N. Geographic differences in species boundaries among members of the Montastraea annularis complex based on molecular and morphological markers. Evolution 2004, 58, 324–337. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Sahl, J.W. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbial. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Lathi, L.; Shetty, S.; Blake, T.; Salojarvi, J. Microbiome R Package. 2017. Available online: http://microbiome.github.io (accessed on 14 September 2020).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 265–270. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical theory of communication. BSTJAN 1948, 27, 379–423, 623–656. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; Mcglinn, D. Vegan: Community Ecology Package. R Package Version 2.5–1. 2018. Available online: https://cran.r-project.org/package=vegan (accessed on 11 August 2020).
- Moitinho-Silva, L.; Nielsen, S.; Amir, A.; Gonzalez, A.; Ackermann, G.L.; Cerrano, C.; Steinert, G. The sponge microbiome project. Gigascience 2017, 6, gix077. [Google Scholar] [CrossRef]
- Dat, T.T.H.; Steinert, G.; Cuc, N.T.K.; Smidt, H.; Sipkema, D. Archaeal and bacterial diversity and community composition from 18 phylogenetically divergent sponge species in Vietnam. Peer J. 2018, 6, e4970. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Laport, M.S.; Pinheiro, U.; Rachid, C.T.C.C. Freshwater Sponge Tubella variabilis Presents Richer Microbiota Than Marine Sponge Species. Front. Microbiol. 2019, 10, 2799. [Google Scholar] [CrossRef]
- Thomas, T.; Moitinho-Silva, L.; Lurgi, M.; Björk, J.R.; Easson, C.; Astudillo-García, C.; Chaves-Fonnegra, A. Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Franco, C.M.; Sorokin, S.J.; Zhang, W. Development of a multilocus-based approach for sponge (phylum Porifera) identification: Refinement and limitations. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Steinert, G.; Busch, K.; Bayer, K.; Kodami, S.; Arbizu, P.M.; Kelly, M.; Millis, S.; Erpenbeck, D.; Dohrmann, M.; Wörheide, G.; et al. Compositional and quantitative insights into bacterial and archaeal communities of South Pacific deep-sea sponges (Demospongiae and Hexactinellida). Front. Microbiol. 2020, 11, 716. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Díez-Vives, C.; Batani, G.; Esteves, A.I.; Jahn, M.T.; Thomas, T. Integrated metabolism in sponge–microbe symbiosis revealed by genome-centered metatranscriptomics. ISME J. 2017, 11, 1651–1666. [Google Scholar] [CrossRef] [PubMed]
- Moeller, F.U.; Webster, N.S.; Herbold, C.W.; Behnam, F.; Domman, D.; Albertsen, M.; Mooshammer, M.; Markert, S.; Turaev, D.; Becher, D.; et al. Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella Basta. Environ. Microbiol. 2019, 21, 3831–3854. [Google Scholar] [CrossRef] [PubMed]
- Sharp, K.H.; Eam, B.; Faulkner, D.J.; Haygood, M.G. Vertical transmission of diverse microbes in the tropical sponge Corticium Appl. Environ. Microbiol. 2007, 73, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Steger, D.; Ettinger-Epstein, P.; Whalan, S.; Hentschel, U.; De Nys, R.; Wagner, M.; Taylor, M.W. Diversity and mode of transmission of ammonia-oxidizing archaea in marine sponges. Environ. Microbiol. 2008, 10, 1087–1094. [Google Scholar] [CrossRef]
- Turon, M.; Uriz, M.J. New insights into the archaeal consortium of tropical sponges. Front. Mar. Sci. 2020, 6. [Google Scholar] [CrossRef]
- Gonçalves, A.C.; Franco, T.; Califano, G.; Dowd, S.E.; Pohnert, G.; Costa, R. Draft genome sequence of Vibrio sp. strain Vb278, an antagonistic bacterium isolated from the marine sponge Sarcotragus spinosulus. Genome Announc. 2015, 3, e00521-15. [Google Scholar] [CrossRef]
- Choudhury, J.D.; Pramanik, A.; Webster, N.S.; Llewellyn, L.E.; Gachhui, R.; Mukherjee, J. Draft Genome Sequence of Pseudoalteromonas sp. Strain NW 4327 (MTCC 11073, DSM 25418), a Pathogen of the Great Barrier Reef Sponge Rhopaloeides odorabile. Genome Announc. 2014, 2, e00001-14. [Google Scholar] [CrossRef]
- Sakai-Kawada, F.E.; Yakym, C.J.; Helmkampf, M.; Hagiwara, K.; Ip, C.G.; Antonio, B.J.; Armstrong, E.; Ulloa, W.J.; Awaya, J.D. Draft genome sequence of marine sponge symbiont Pseudoalteromonas luteoviolacea IPB1, isolated from Hilo, Hawaii. Gen. Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Arnosti, C. Microbial extracellular enzymes and the marine carbon cycle. Ann. Rev. Mar. Sci. 2011, 3, 401–425. [Google Scholar] [CrossRef] [PubMed]
- Arnosti, C.; Wietz, M.; Brinkhoff, T.; Hehemann, J.H.; Probandt, D.; Zeugner, L.; Amann, R. The biogeochemistry of marine polysaccharides: Sources, inventories, and bacterial drivers of the carbohydrate cycle. Annu. Rev. Mar. Sci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.T.; Lee, J. Global distribution and prevalence of Arcobacter in food and water. Zoonoses Public Health. 2015, 62, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Fera, M.T.; Maugeri, T.L.; Gugliandolo, C.; Beninati, C.; Giannone, M.; La Camera, E.; Carbone, M. Detection of Arcobacter spp. in the coastal environment of the Mediterranean Sea. Appl. Environ. Microbiol. 2004, 70, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, T.L.; Irrera, G.P.; Lentini, V.; Carbone, M.; Fera, M.T.; Gugliandolo, C. Detection and enumeration of Arcobacter spp. in the coastal environment of the Straits of Messina (Italy). New Microbiol. 2005, 28, 177–182. [Google Scholar]
- Collado, L.; Inza, I.; Guarro, J.; Figueras, M.J. Presence of Arcobacter spp. in environmental waters correlates with high levels of fecal pollution. Environ. Microbiol. 2008, 10, 1635–1640. [Google Scholar] [CrossRef]
- Fera, M.T.; Gugliandolo, C.; Lentini, V.; Favaloro, A.; Bonanno, D.; La Camera, E.; Maugeri, T.L. Specific detection of Arcobacter spp. in estuarine waters of Southern Italy by PCR and fluorescent in situ hybridization. Lett. Appl. Microbiol. 2010, 50, 65–70. [Google Scholar] [CrossRef]
- Diéguez, A.L.; Romalde, J.L. Complete genome sequence of Arcobacter sp. strain LFT 1.7 isolated from Great Scallop (Pecten maximus) Larvae. Genome Announc. 2017, 5, e01617-16. [Google Scholar] [CrossRef]
- García-Bonilla, E.; Brandão, P.F.; Pérez, T.; Junca, H. Stable and enriched Cenarchaeum symbiosum and uncultured Betaproteobacteria HF1 in the microbiome of the Mediterranean sponge Haliclona fulva (Demospongiae: Haplosclerida). Microb. Ecol. 2018, 77, 25–36. [Google Scholar] [CrossRef]
- Cárdenas, C.A.; González-Aravena, M.; Font, A.; Hestetun, J.T.; Hajdu, E.; Trefault, N.; Malmberg, M.; Bongcam-Rudloff, E. High similarity in the microbiota of cold-water sponges of the Genus Mycale from two different geographical areas. PeerJ 2018, 6, e4935. [Google Scholar] [CrossRef] [PubMed]
- Balvočiūtė, M.; Huson, D.H. SILVA, RDP, Greengenes, NCBI and OTT—how do these taxonomies compare? BMC Genomics. 2017, 18, 1–8. [Google Scholar] [CrossRef]
- Almeida, R.; Mitchell, A.L.; Tarkowska, A.; Finn, R.D. Benchmarking taxonomic assignments based on 16S rRNA gene profiling of the microbiota from commonly sampled environments. GigaScience. 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Blanquer, A.; Uriz, M.J.; Galand, P.E. Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ. Microbiol. 2013, 15, 3008–3019. [Google Scholar] [CrossRef]
- de Caralt, S.; Uriz, M.J.; Ereskovsky, A.V.; Wijffels, R.H. Embryo development of Corticium candelabrum (Demospongiae: Homosclerophorida). Invertebr. Biol. 2007, 126, 211–219. [Google Scholar] [CrossRef]
- Maldonado, M.; Riesgo, A. Reproductive output in a Mediterranean population of the homosclerophorid Corticium candelabrum (Porifera, Demospongiae), with notes on the ultrastructure and behavior of the larva. Mar. Ecol. 2008, 29, 298–316. [Google Scholar] [CrossRef]
- Gloeckner, V.; Hentschel, U.; Ereskovsky, A.V.; Schmitt, S. Unique and species-specific microbial communities in Oscarella lobularis and other Mediterranean Oscarella species (Porifera: Homoscleromorpha). Mar. Biol. 2013, 160, 781–791. [Google Scholar] [CrossRef]
- Moitinho-Silva, L.; Steinert, G.; Nielsen, S.; Hardoim, C.C.P.; Wu, Y.-C.; López-Legentil, S.; Marchant, R.; Webster, N.; Thomas, T.; Hentschel, U. Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 2017, 8, 752. [Google Scholar] [CrossRef]
- Lurgi, M.; Thomas, T.; Wemheuer, B.; Webster, N.S.; Montoya, J.M. Modularity and predicted functions of the global sponge-microbiome network. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Cleary, D.F.R.; Swierts, T.; Coelho, F.J.R.C.; Polónia, A.R.M.; Huang, Y.M.; Ferreira, M.R.S.; Putchakarn, S.; Carvalheiro, L.; van der Ent, E.; Ueng, J.P.; et al. The sponge microbiome within the greater coral reef microbial metacommunity. Nat. Commun. 2019, 10, 1644. [Google Scholar] [CrossRef]
- Vacelet, J.; Donadey, C. Electron microscope study of the association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 1977, 30, 301–314. [Google Scholar] [CrossRef]
- Hentschel, U.; Usher, K.M.; Taylor, M.W. Marine sponges as microbial fermenters. FEMS Microbiol. Ecol. 2006, 55, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Weisz, J.B.; Lindquist, N.; Martens, C.S. Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia 2008, 155, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Weisz, J.B.; Hentschel, U.; Lindquist, N.; Martens, C.S. Linking abundance and diversity of sponge-associated microbial communities to metabolic differences in host sponges. Mar. Biol. 2007, 152, 475–483. [Google Scholar] [CrossRef]
- DeLong, E.F.; Wu, K.Y.; Prézelin, B.B.; Jovine, R.V. High abundance of Archaea in Antarctic marine picoplankton. Nature 1994, 371, 695–697. [Google Scholar] [CrossRef]
- Stocker, R. Marine microbes see a sea of gradients. Science 2012, 338, 628–633. [Google Scholar] [CrossRef]
- Taylor, M.W.; Schupp, P.J.; Dahllöf, I.; Kjelleberg, S.; Steinberg, P.D. Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environ. Microbiol. 2004, 6, 121–130. [Google Scholar] [CrossRef]
- Webster, N.S.; Negri, A.P.; Munro, M.M.; Battershill, C.N. Diverse microbial communities inhabit Antarctic sponges. Environ. Microbiol. 2004, 6, 288–300. [Google Scholar] [CrossRef]
- Giles, E.C.; Kamke, J.; Moitinho-Silva, L.; Taylor, M.W.; Hentschel, U.; Ravasi, T.; Schmitt, S. Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 2013, 83, 232–241. [Google Scholar] [CrossRef]
- Gloeckner, V.; Wehrl, M.; Moitinho-Silva, L.; Gernert, C.; Schupp, P.; Pawlik, J.R.; Lindquist, N.L.; Erpenbeck, D.; Wörheide, G.; Hentschel, U. The HMA-LMA dichotomy revisited: An electron microscopical survey of 56 sponge species. Biol. Bull. 2014, 227, 78–88. [Google Scholar] [CrossRef]
- Ribes, M.; Dziallas, C.; Coma, R.; Riemann, L. Microbial diversity and putative diazotrophy in high- and low-abundance mediterranean sponges. Appl. Env. Microbiol. 2015, 81, 5683–5693. [Google Scholar] [CrossRef] [PubMed]
- Ribes, M.; Calvo, E.; Movilla, J.; Logares, R.; Coma, R.; Pelejero, C. Restructuring of the sponge microbiome favors tolerance to ocean acidification. Environ. Microbiol. Rep. 2016, 8, 536–544. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, B.F.R.; Lopes, I.R.; Canellas, A.L.B.; Muricy, G.; Dobson, A.D.W.; Laport, M.S. Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea. Microorganisms 2020, 8, 1978. https://doi.org/10.3390/microorganisms8121978
Oliveira BFR, Lopes IR, Canellas ALB, Muricy G, Dobson ADW, Laport MS. Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea. Microorganisms. 2020; 8(12):1978. https://doi.org/10.3390/microorganisms8121978
Chicago/Turabian StyleOliveira, Bruno F. R., Isabelle R. Lopes, Anna L. B. Canellas, Guilherme Muricy, Alan D. W. Dobson, and Marinella S. Laport. 2020. "Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea" Microorganisms 8, no. 12: 1978. https://doi.org/10.3390/microorganisms8121978
APA StyleOliveira, B. F. R., Lopes, I. R., Canellas, A. L. B., Muricy, G., Dobson, A. D. W., & Laport, M. S. (2020). Not That Close to Mommy: Horizontal Transmission Seeds the Microbiome Associated with the Marine Sponge Plakina cyanorosea. Microorganisms, 8(12), 1978. https://doi.org/10.3390/microorganisms8121978