Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core


 , ,
, ,  and
and 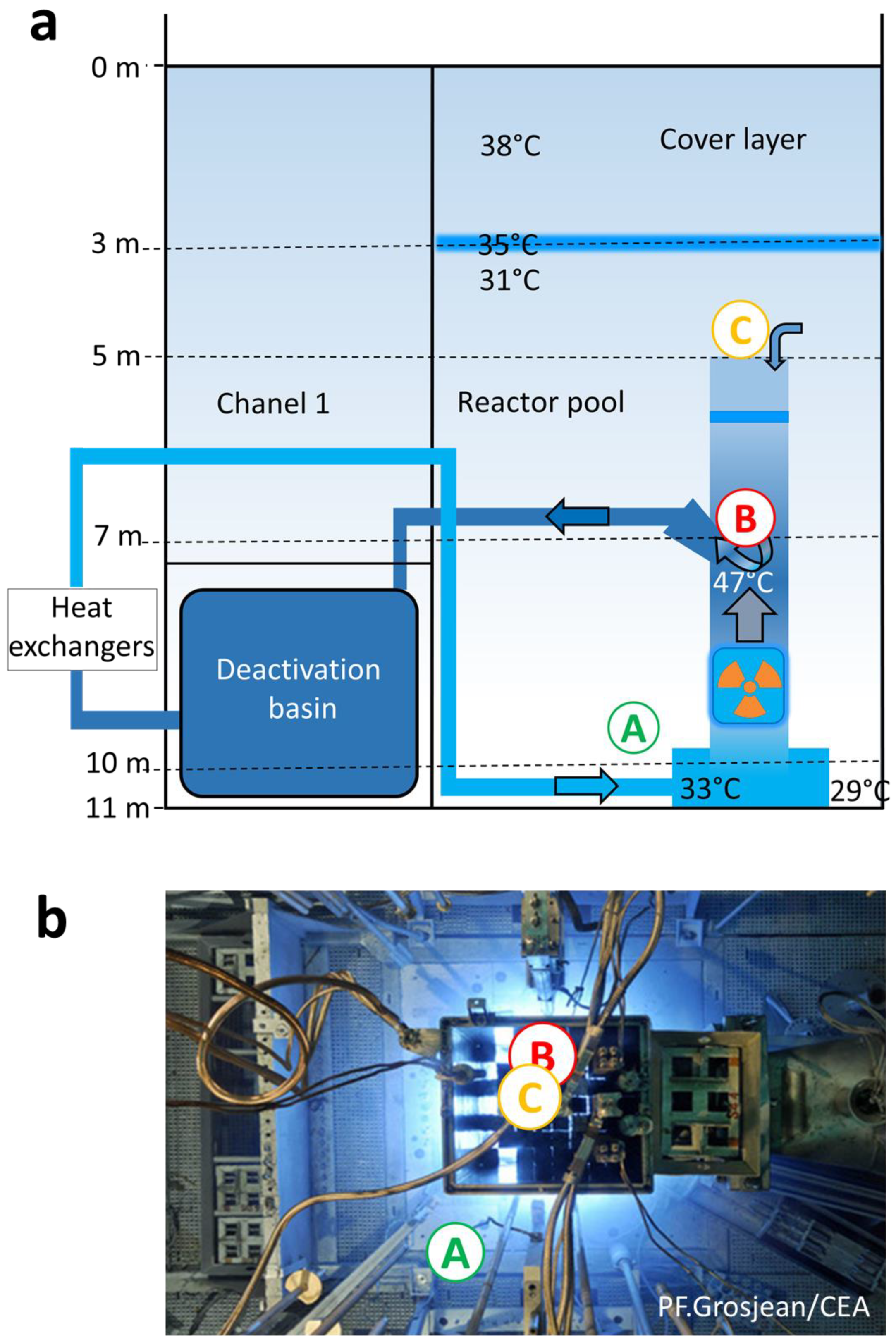{kind=link}
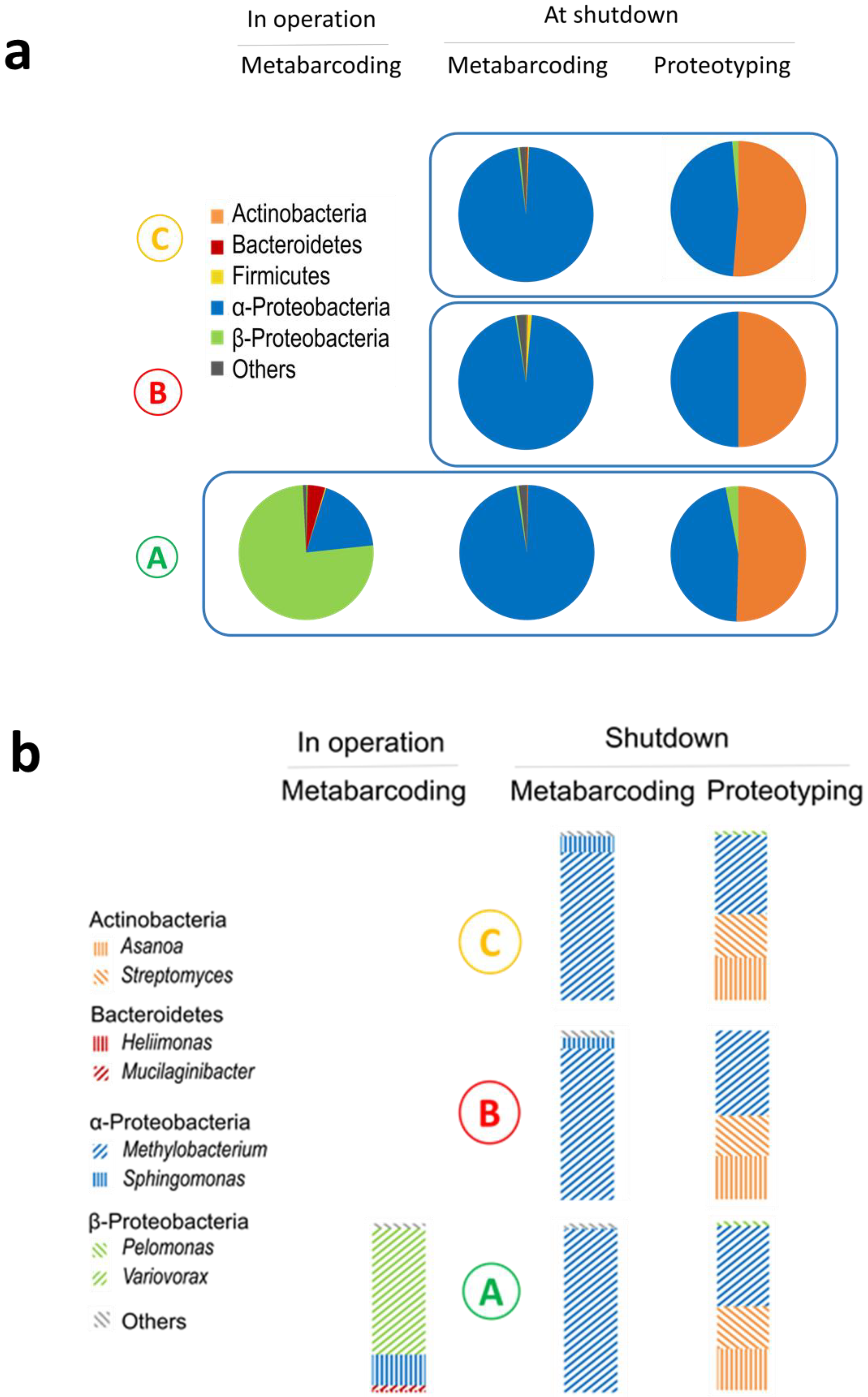{kind=link}
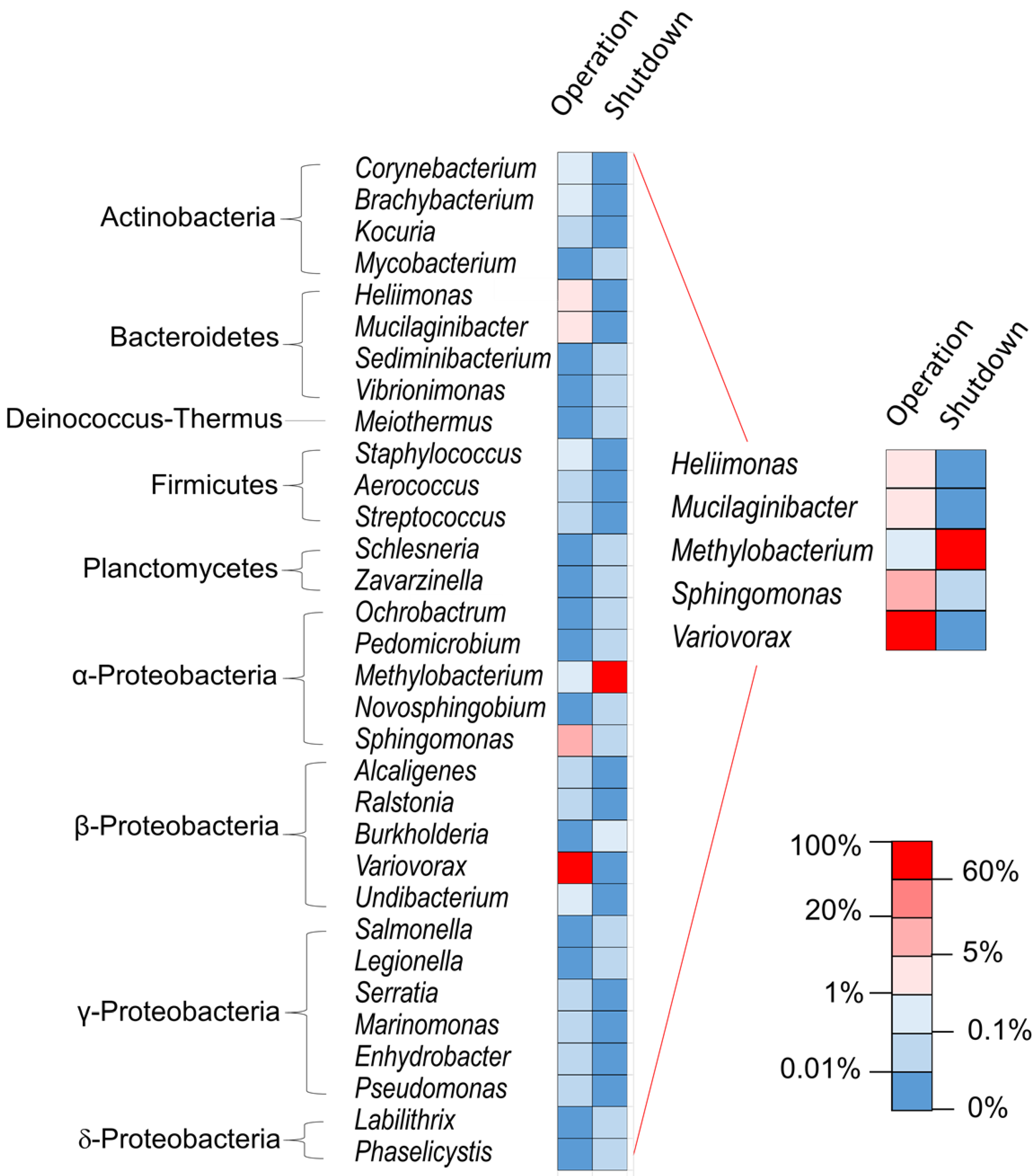{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Water Samples from the Cooling Pool of the Nuclear Reactor Core
2.2. DNA Extraction, Illumina Sequencing, and Sequence Data Analysis by Metabarcoding
2.3. Protein Extraction, Peptide Analysis Using nanoLC–MS/MS, and Taxonomic Analysis by Proteotyping
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Chicote, E.; Garcia, A.M.; Moreno, D.A.; Sarro, M.I.; Lorenzo, P.I.; Montero, F. Isolation and identification of bacteria from spent nuclear fuel pools. J. Ind. Microbiol. Biotechnol. 2005, 32, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Sarro, M.I.; Garcia, A.M.; Moreno, D.A. Biofilm formation in spent nuclear fuel pools and bioremediation of radioactive water. Int. Microbiol. 2005, 8, 223–230. [Google Scholar] [PubMed]
- Masurat, P.; Fru, E.C.; Pedersen, K. Identification of Meiothermus as the dominant genus in a storage system for spent nuclear fuel. J. Appl. Microbiol. 2005, 98, 727–740. [Google Scholar] [CrossRef]
- Rivasseau, C.; Farhi, E.; Compagnon, E.; de Gouvion Saint Cyr, D.; van Lis, R.; Falconet, D.; Kuntz, M.; Atteia, A.; Couté, A. Coccomyxa actinabiotis sp. nov. (Trebouxiophyceae; Chlorophyta); a new green microalga living in the spent fuel cooling pool of a nuclear reactor. J. Phycol. 2016, 52, 689–703. [Google Scholar] [CrossRef]
- Rivasseau, C.; Farhi, E.; Atteia, A.; Couté, A.; Gromova, M.; de Gouvion Saint Cyr, D.; Boisson, A.-M.; Féret, A.-S.; Compagnon, E.; Bligny, R. An extremely radioresistant green eukaryote for radionuclide bio-decontamination in the nuclear industry. Energy Environ. Sci. 2013, 6, 1230. [Google Scholar] [CrossRef]
- Stokell, J.R.; Steck, T.R. Viable but Nonculturable Bacteria. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.P.; Edwards, D.J.; Harwich, M.D., Jr.; Rivera, M.C.; Fettweis, J.M.; Serrano, M.G.; Reris, R.A.; Sheth, N.U.; Huang, B.; Girerd, P.; et al. The truth about metagenomics: Quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015, 15, 66. [Google Scholar] [CrossRef]
- Grenga, L.; Pible, O.; Armengaud, J. Pathogen proteotyping: A rapidly developing application of mass spectrometry to adress clinical concerns. Clin. Mass. Spectrom. 2019. [Google Scholar] [CrossRef]
- Kleiner, M.; Thorson, E.; Sharp, C.E.; Dong, X.; Liu, D.; Li, C.; Strous, M. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 2017, 8, 1558. [Google Scholar] [CrossRef]
- Report CEA/DEN/DANS/11-42. INB 40-Réacteurs OSIRIS et ISIS-Evaluation Complémentaire de la Sûreté au Regard de L’accident Survenu à la Centrale de Fukushima Daiichi. 2011. Available online: https://www.asn.fr/sites/rapports-exploitants-ecs/CEA/CEA-Osiris.pdf (accessed on 24 November 2020).
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87–98. [Google Scholar] [CrossRef]
- Goffau, M.; Lager, S.; Salter, S.J.; Wagner, J.; Kronbichler, A.; Charnock-Jones, D.S.; Peacock, S.J.; Smith, G.C.S.; Parkhill, J. Recognizing the reagent microbiome. Nature Microbiol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Vilchez-Vargas, R.; Geffers, R.; Suárez-Diez, M.; Conte, I.; Waliczek, A.; Kaser, V.S.; Kralova, M.; Junca, H.; Pieper, D.H. Analysis of the microbial gene landscape and transcriptome for aromatic pollutants and alkane degradation using a novel internally calibrated microarray system. Environ. Microbiol. 2013, 15, 1016–1039. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities; time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Mysara, M.; Njima, M.; Leys, N.; Raes, J.; Monsieurs, P. From reads to operational taxonomic units: An ensemble processing pipeline for MiSeq amplicon sequencing data. GigaScience 2017, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B. Introducing mothur: Open-source; platform-independent; community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Hartmann, E.M.; Allain, F.; Gaillard, J.C.; Pible, O.; Armengaud, J. Taking the shortcut for high-throughput shotgun proteomic analysis of bacteria. Methods Mol. Biol. 2014, 1197, 275–285. [Google Scholar]
- Clair, G.; Armengaud, J.; Duport, C. Restricting fermentative potential by proteome remodeling: An adaptive strategy evidenced in Bacillus cereus. Mol. Cell. Proteom. 2012, 11, M111.013102. [Google Scholar] [CrossRef]
- Klein, G.; Mathé, C.; Biola-Clier, M.; Devineau, S.; Drouineau, E.; Hatem, E.; Marichal, L.; Alonso, B.; Gaillard, J.-C.; Lagniel, G.; et al. RNA-binding proteins are a major target of silica nanoparticles in cell extracts. Nanotoxicology 2016, 10, 1555–1564. [Google Scholar] [CrossRef]
- Pible, O.; Allain, F.; Jouffret, V.; Culotta, K.; Miotello, G.; Armengaud, J. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics”. Microbiome 2020, 8, 30. [Google Scholar] [CrossRef]
- Bagwell, C.E.; Noble, P.A.; Milliken, C.E.; Li, D.; Kaplan, D.I. Amplicon Sequencing Reveals Microbiological Signatures in Spent Nuclear Fuel Storage Basins. Front. Microbiol. 2018, 9, 377. [Google Scholar] [CrossRef]
- Hayoun, K.; Pible, O.; Petit, P.; Allain, P.; Jouffret, V.; Culotta, K.; Rivasseau, C.; Armengaud, J.; Alpha-Bazin, B. Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool. Microorganisms 2020, 8, 1525. [Google Scholar] [CrossRef]
- Mappa, C. Exploration de Nouveaux Concepts pour les Analyses Quantitatives et Fonctionnelles de Microbiotes Modèles D’intérêt Dual. Ph.D. Thesis, Université de Montpellier, Montpellier, France, 2018. [Google Scholar]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E.; et al. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Petit, P. Analyse des Espèces Biologiques Vivant dans les Piscines des Installations Nucléaires: Vers L’identification D’espèces Radiorésistantes. Ph.D. Thesis, Université Grenoble-Alpes, Grenoble, France, 2018. [Google Scholar]
- Davis, D.H.; Doudoroff, M.; Stanier, R.Y.; Mandel, M. Proposal to reject the genus Hydrogenomonas: Taxonomic implications. Int. J. Syst. Evol. Microbiol. 1969, 19, 375–390. [Google Scholar] [CrossRef]
- Brenner, D.J.; Krieg, N.R.; Staley, J.T. Bergey’s Manual of Systematic Bacteriology—The Alpha-; Beta-; Delta- and Epsilonproteobacteria. 2005. Available online: https://www.researchgate.net/publication/256294896_Bergey%27s_Manual_of_Systematic_Bacteriology_Vol_2_Part_C_The_Alpha-_Beta-_Delta-_and_Epsilonproteobacteria (accessed on 24 November 2020).
- Yan, X.; Luo, X.; Zhao, M. Metagenomic analysis of microbial community in uranium-contaminated soil. Appl. Microbiol. Biotechnol. 2016, 100, 299–310. [Google Scholar] [CrossRef]
- Han, J.-I.; Choi, H.-K.; Lee, S.-W.; Orwin, P.M.; Kim, J.; LaRoe, S.L.; Kim, T.-G.; O’Neil, J.; Leadbetter, J.R.; Lee, S.Y.; et al. Complete Genome Sequence of the Metabolically Versatile Plant Growth-Promoting Endophyte Variovorax paradoxus. J. Bacteriol. 2011, 193, 1183–1190. [Google Scholar] [CrossRef]
- Chapon, V.; Piette, L.; Vesvres, M.H.; Coppin, F.; Marrec, C.L.; Christen, R.; Theodorakopoulos, N.; Février, L.; Levchuk, S.; Martin-Garin, A.; et al. Microbial diversity in contaminated soils along the T22 trench of the Chernobyl experimental platform. Appl. Geochem. 2012, 27, 1375–1383. [Google Scholar] [CrossRef]
- Mao, J.; Tang, Q.; Zhang, Z.; Wang, W.; Wei, D.; Huang, Y.; Liu, Z.; Shi, Y.; Goodfellow, M. Streptomyces radiopugnans sp. nov.; a radiation-resistant actinomycete isolated from radiation-polluted soil in China. Int. J. Syst. Evol. Microbiol. 2012, 57, 2578–2582. [Google Scholar] [CrossRef]
- Nogueira, F.; Botelho, M.L.; Tenreiro, R. Radioresistance studies in Methylobacterium spp. Radiat. Phys. Chem. 1998, 52, 3. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petit, P.C.M.; Pible, O.; Eesbeeck, V.V.; Alban, C.; Steinmetz, G.; Mysara, M.; Monsieurs, P.; Armengaud, J.; Rivasseau, C. Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core. Microorganisms 2020, 8, 1857. https://doi.org/10.3390/microorganisms8121857
Petit PCM, Pible O, Eesbeeck VV, Alban C, Steinmetz G, Mysara M, Monsieurs P, Armengaud J, Rivasseau C. Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core. Microorganisms. 2020; 8(12):1857. https://doi.org/10.3390/microorganisms8121857
Chicago/Turabian StylePetit, Pauline C. M., Olivier Pible, Valérie Van Eesbeeck, Claude Alban, Gérard Steinmetz, Mohamed Mysara, Pieter Monsieurs, Jean Armengaud, and Corinne Rivasseau. 2020. "Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core" Microorganisms 8, no. 12: 1857. https://doi.org/10.3390/microorganisms8121857
APA StylePetit, P. C. M., Pible, O., Eesbeeck, V. V., Alban, C., Steinmetz, G., Mysara, M., Monsieurs, P., Armengaud, J., & Rivasseau, C. (2020). Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core. Microorganisms, 8(12), 1857. https://doi.org/10.3390/microorganisms8121857