Global Proteomic Profiling of Piscirickettsia salmonis and Salmon Macrophage-Like Cells during Intracellular Infection

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. SHK-1 Cell Culture Infection Assays
2.3. Global Proteomic Profiling Using Q-Exactive Mass Spectrometry
2.4. Bioinformatics Analysis of Proteins Detected by Global Proteomic Profiling
2.5. Bacterial RNA Purification and Transcripts Quantification
3. Results
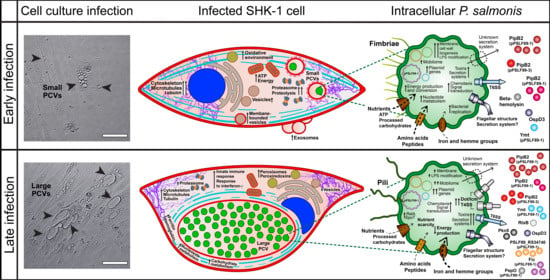
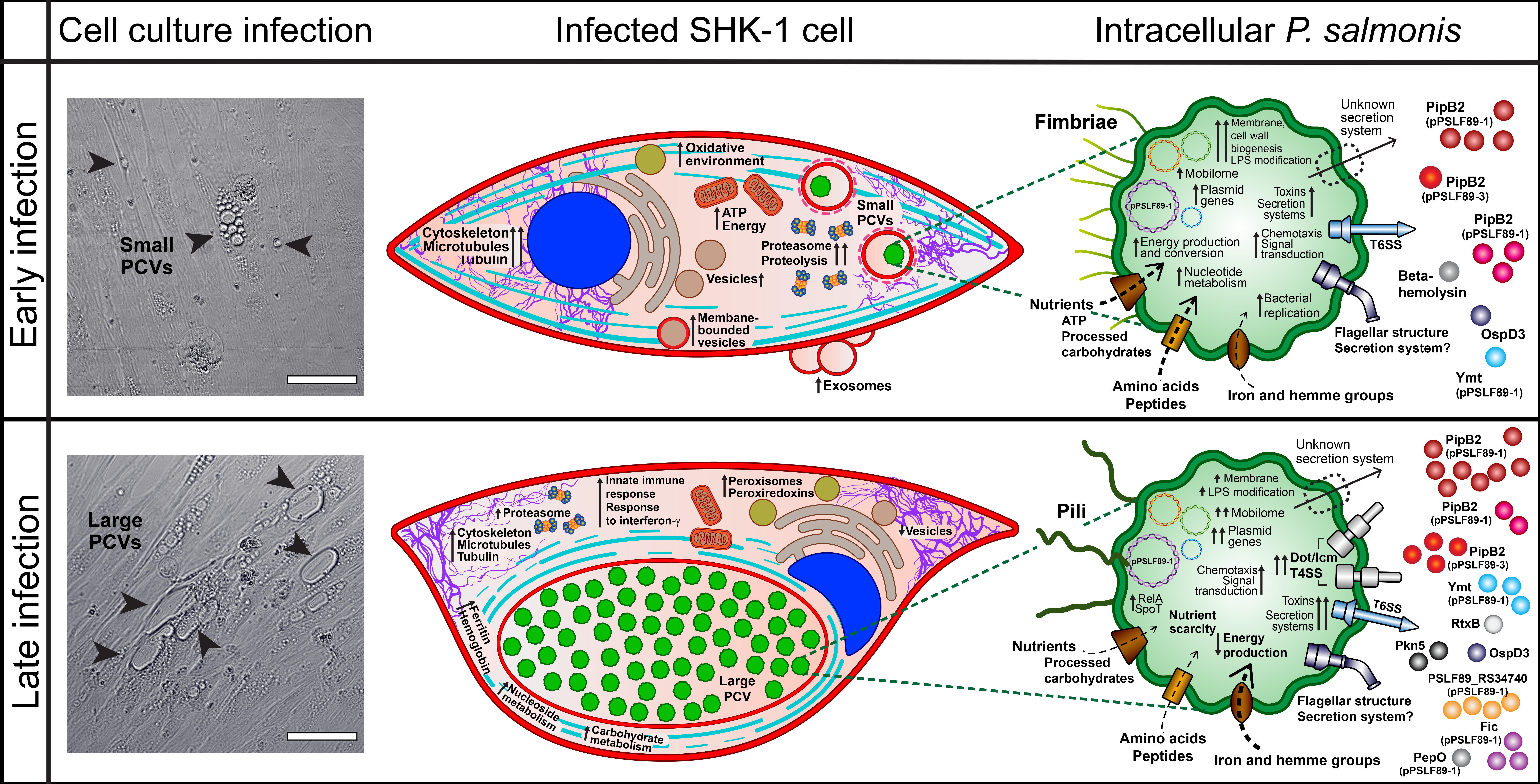
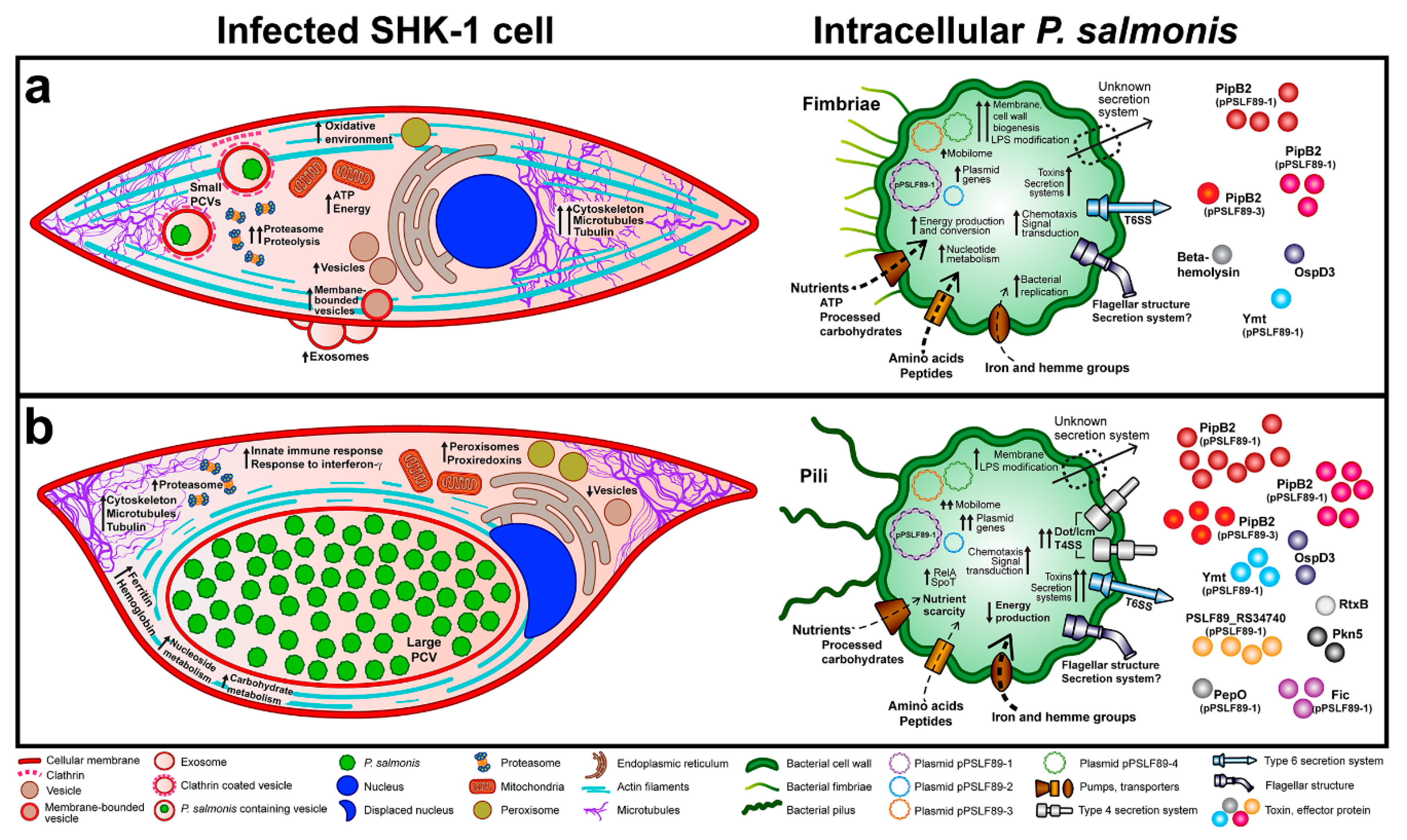
3.1. P. salmonis Infection Progress in Fish Macrophages Culture
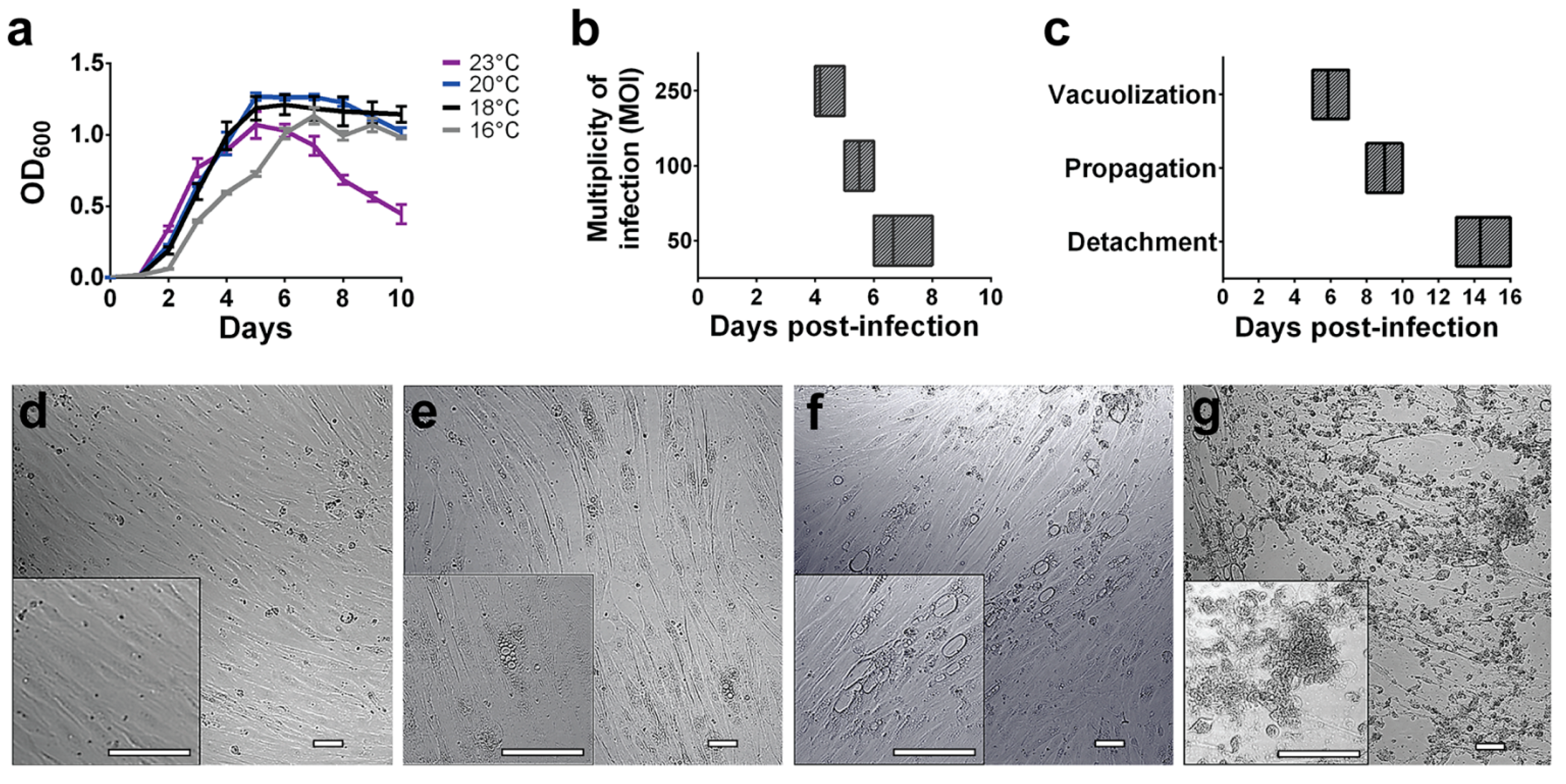
3.2. Global Proteomic Profiling of P. salmonis Infection in S. salar SHK-1 Cell Cultures
3.2.1. P. salmonis Proteome
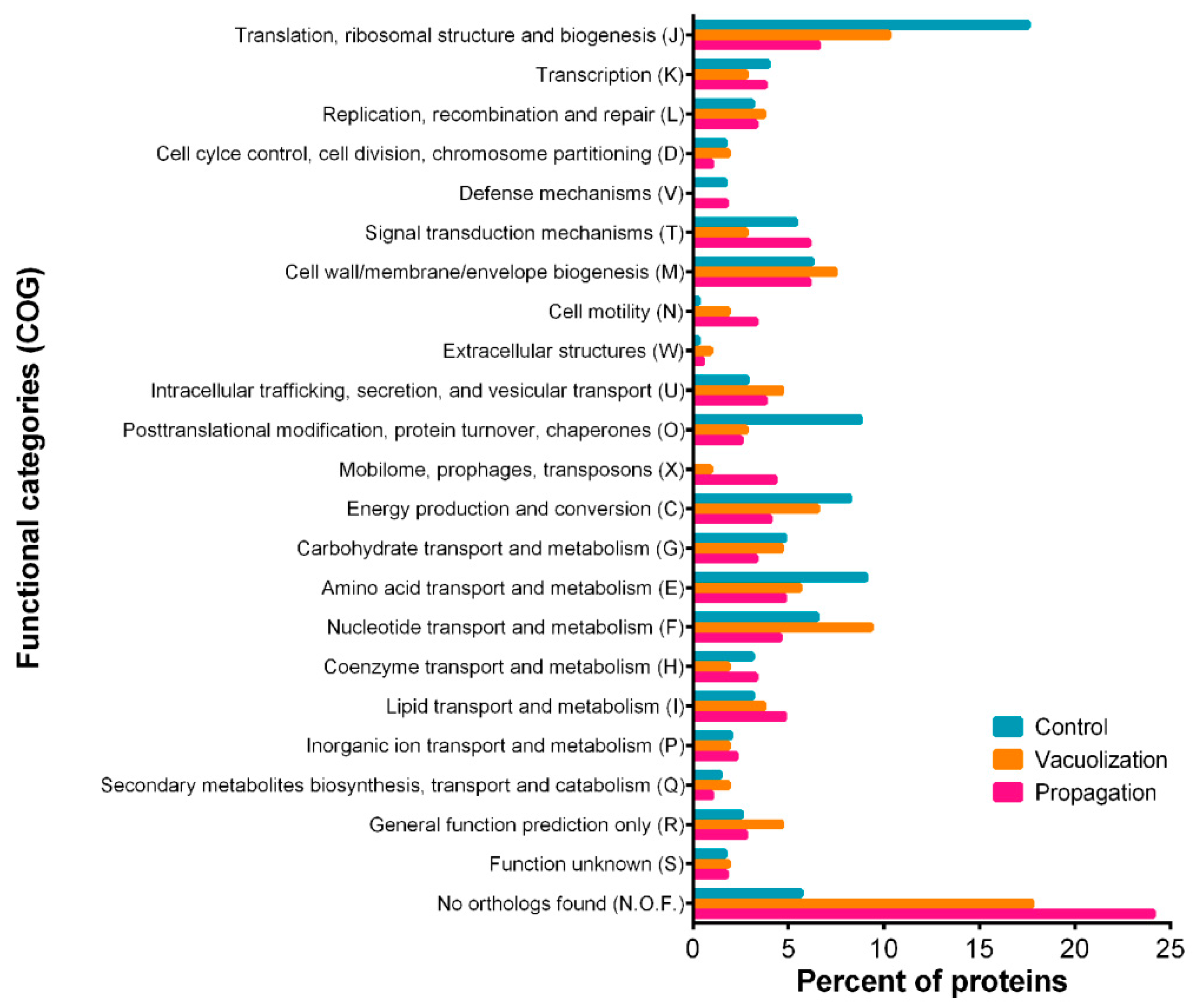
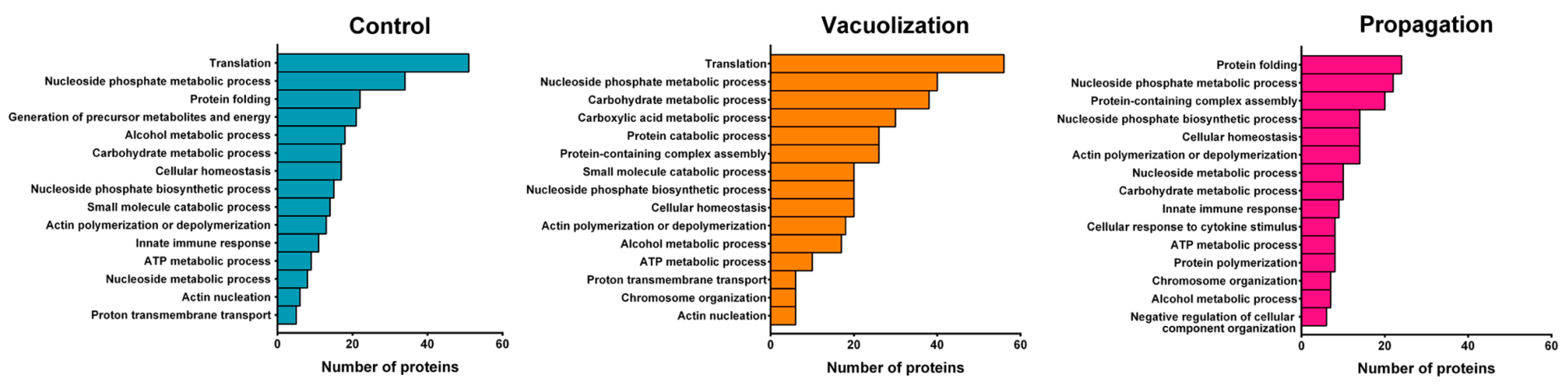
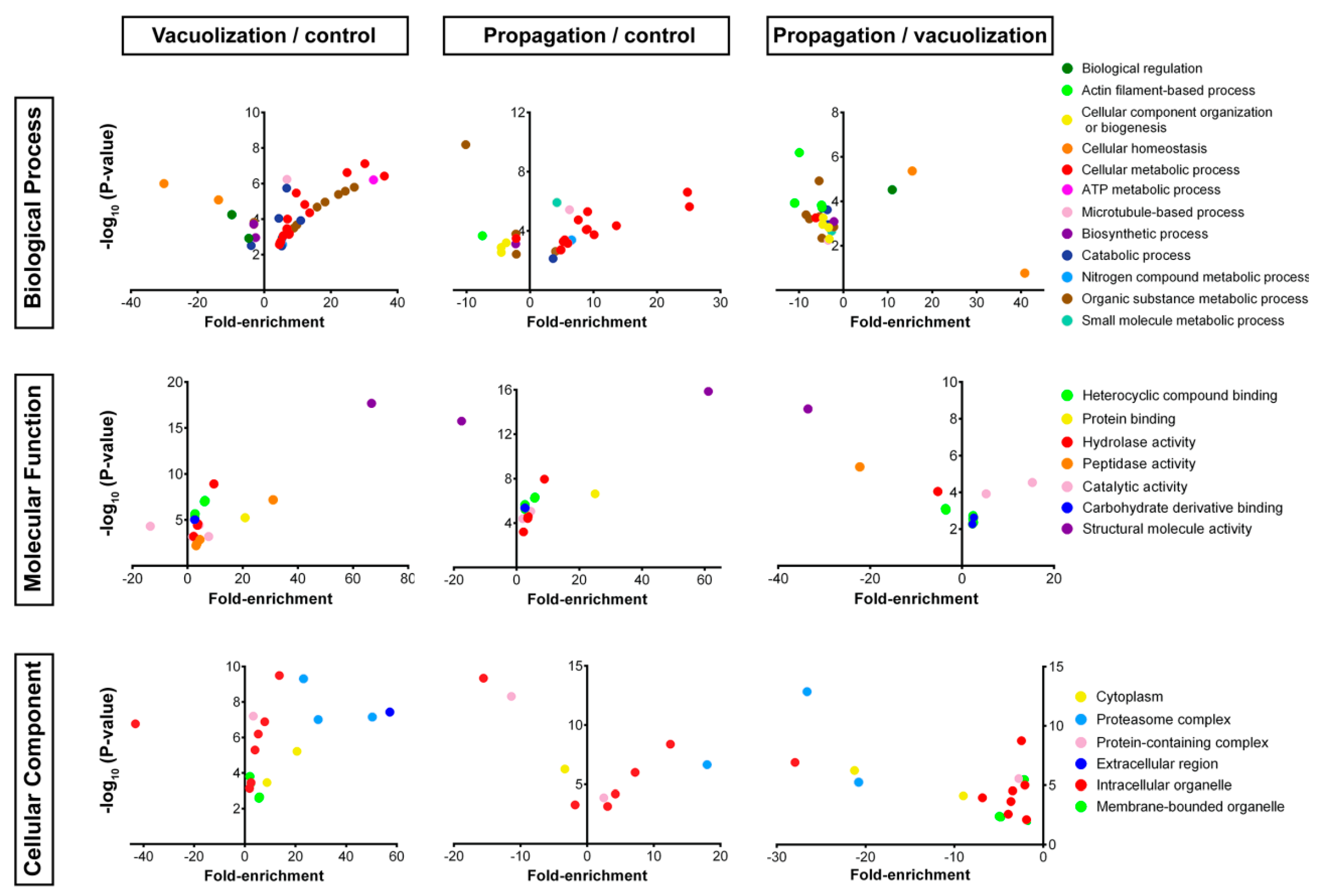
3.2.2. S. salar SHK-1 Cells Proteome
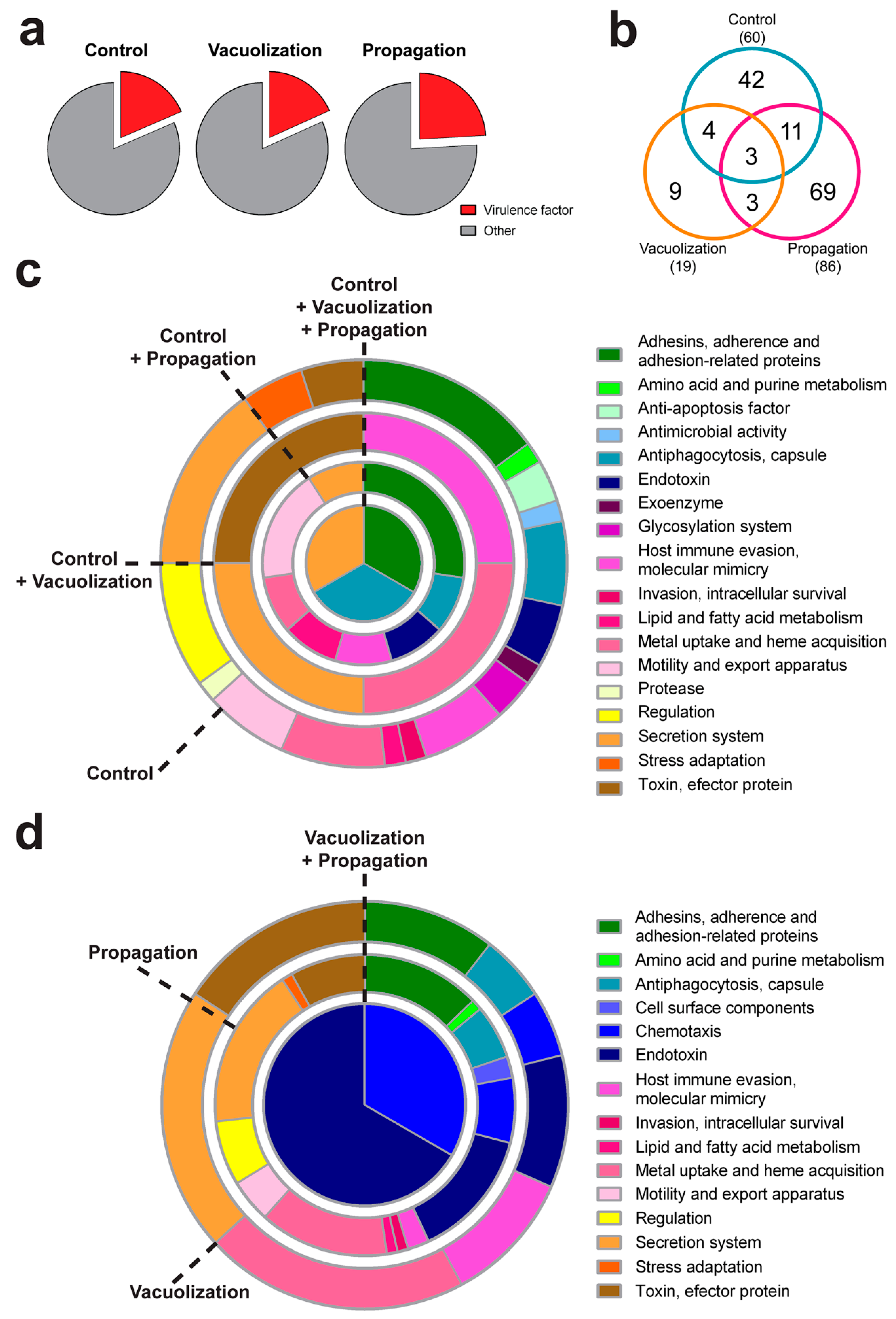
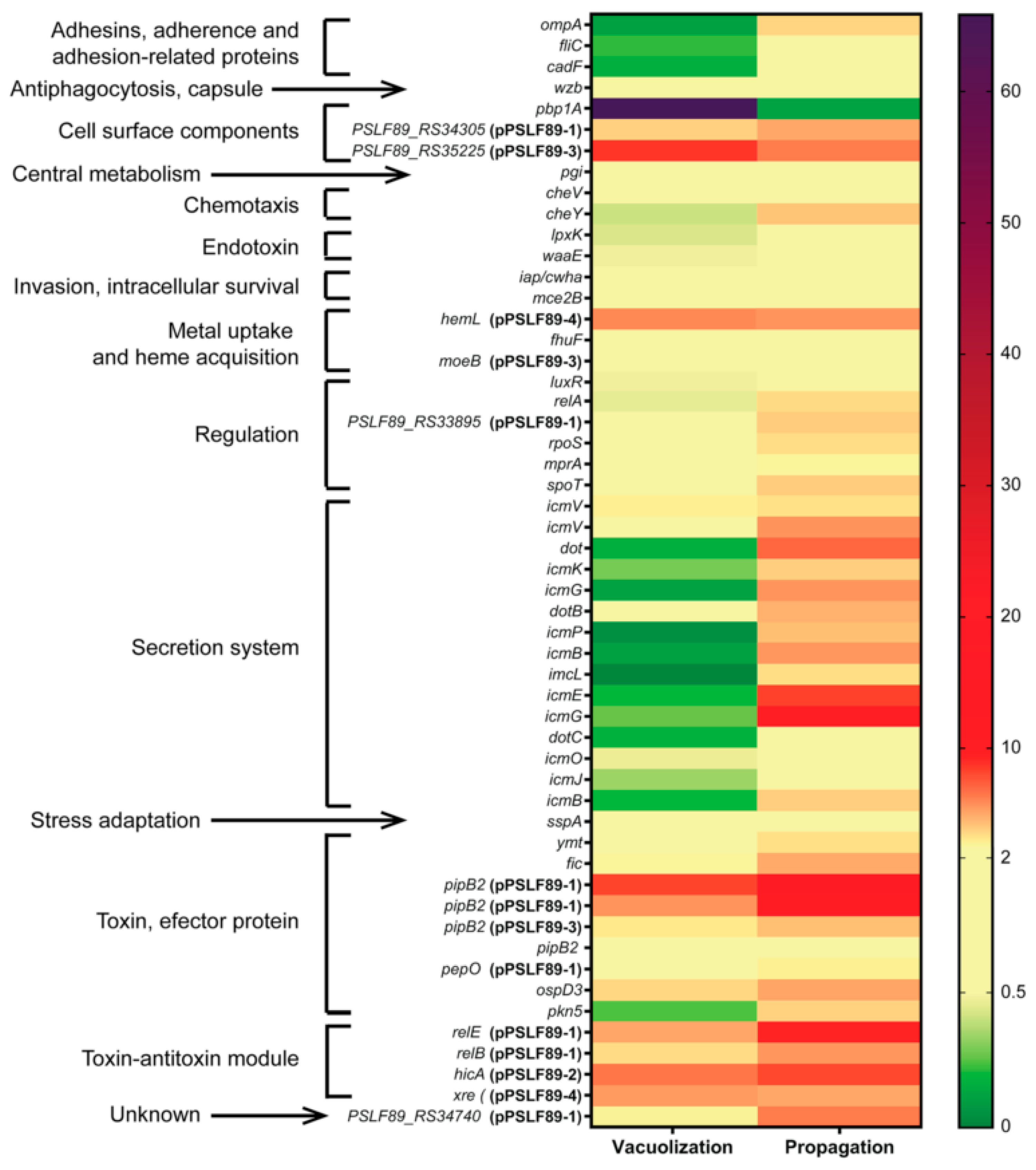
3.3. P. salmonis Virulence Factors
3.4. Gene Expression of Selected P. salmonis Virulence Factors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotter, P.A.; DiRita, V.J. Bacterial virulence gene regulation: An evolutionary perspective. Annu. Rev. Microbiol. 2000, 54, 519–565. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Rozas, M.; Enriquez, R. Piscirickettsiosis and Piscirickettsia salmonis in fish: A review. J. Fish Dis. 2014, 37, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Makrinos, D.L.; Bowden, T.J. Growth characteristics of the intracellular pathogen, Piscirickettsia salmonis, in tissue culture and cell-free media. J. Fish Dis. 2017, 40, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Mancilla, M.; Saavedra, J.; Grandón, M.; Tapia, E.; Navas, E.; Grothusen, H.; Bustos, P. The mutagenesis of a type IV secretion system locus of Piscirickettsia salmonis leads to the attenuation of the pathogen in Atlantic salmon, Salmo salar. J. Fish Dis. 2018, 41, 625–634. [Google Scholar] [CrossRef]
- Saavedra, J.; Grandón, M.; Villalobos-González, J.; Bohle, H.; Bustos, P.; Mancilla, M. Isolation, functional characterization and transmissibility of p3PS10, a multidrug resistance plasmid of the fish pathogen Piscirickettsia salmonis. Front. Microbiol. 2018, 9, 923. [Google Scholar] [CrossRef]
- Tacchi, L.; Bron, J.E.; Taggart, J.B.; Secombes, C.J.; Bickerdike, R.; Adler, M.A.; Takle, H.; Martin, S.A.M. Multiple tissue transcriptomic responses to Piscirickettsia salmonis in Atlantic salmon (Salmo salar). Physiol. Genom. 2011, 43, 1241–1254. [Google Scholar] [CrossRef]
- Rise, M.L.M.; Jones, S.S.R.M.; Brown, G.D.; von Schalburg, K.R.; Davidson, W.S.; Koop, B.F. Microarray analyses identify molecular biomarkers of Atlantic salmon macrophage and hematopoietic kidney response to Piscirickettsia salmonis infection. Physiol. Genom. 2004, 20, 21–35. [Google Scholar] [CrossRef]
- Pulgar, R.; Hödar, C.; Travisany, D.; Zuñiga, A.; Domínguez, C.; Maass, A.; González, M.; Cambiazo, V. Transcriptional response of Atlantic salmon families to Piscirickettsia salmonis infection highlights the relevance of the iron-deprivation defence system. BMC Genom. 2015, 16, 495. [Google Scholar] [CrossRef]
- Fuentealba, P.; Aros, C.; Latorre, Y.; Martinez, I.; Marshall, S.; Ferrer, P.; Albiol, J.; Altamirano, C. Genome-scale metabolic reconstruction for the insidious bacterium in aquaculture Piscirickettsia salmonis. Bioresour. Technol. 2017, 223, 105–114. [Google Scholar] [CrossRef]
- Zúñiga, A.; Aravena, P.; Pulgar, R.; Travisany, D.; Ortiz-Severín, J.; Chávez, F.P.; Maass, A.; González, M.; Cambiazo, V. Transcriptomic changes of Piscirickettsia salmonis during intracellular growth in a salmon macrophage-like cell line. Front. Cell. Infect. Microbiol. 2020, 9, 426. [Google Scholar] [CrossRef] [PubMed]
- Rozas-Serri, M.; Peña, A.; Maldonado, L. Transcriptomic profiles of post-smolt Atlantic salmon challenged with Piscirickettsia salmonis reveal a strategy to evade the adaptive immune response and modify cell-autonomous immunity. Dev. Comp. Immunol. 2018, 81, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Machuca, A.; Martinez, V. Transcriptome analysis of the intracellular facultative pathogen Piscirickettsia salmonis: Expression of putative groups of genes associated with virulence and iron metabolism. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Valenzuela-Miranda, D.; Gallardo-Escárate, C. Dual RNA-Seq uncovers metabolic amino acids dependency of the intracellular bacterium Piscirickettsia salmonis infecting Atlantic salmon. Front. Microbiol. 2018, 9, 2877. [Google Scholar] [CrossRef] [PubMed]
- Jean Beltran, P.M.; Federspiel, J.D.; Sheng, X.; Cristea, I.M. Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol. Syst. Biol. 2017, 13, 922. [Google Scholar] [CrossRef]
- Cortés, M.; Sánchez, P.; Ruiz, P.; Haro, R.; Sáez, J.; Sánchez, F.; Hernández, M.; Oliver, C.; Yáñez, A.J. In vitro expression of Sec-dependent pathway and type 4B secretion system in Piscirickettsia salmonis. Microb. Pathog. 2017, 110, 586–593. [Google Scholar] [CrossRef]
- Sánchez, P.; Oliver, C.; Hernández, M.; Cortés, M.; Cecilia Rauch, M.; Valenzuela, K.; Garduño, R.A.; Avendaño-Herrera, R.; Yáñez, A.J. In vitro genomic and proteomic evidence of a type IV pili-like structure in the fish pathogen Piscirickettsia salmonis. FEMS Microbiol. Lett. 2018, 365, fny169. [Google Scholar] [CrossRef]
- Oliver, C.; Hernández, M.A.; Tandberg, J.I.; Valenzuela, K.N.; Lagos, L.X.; Haro, R.E.; Sánchez, P.; Ruiz, P.A.; Sanhueza-Oyarzún, C.; Cortés, M.A.; et al. The proteome of biologically active membrane vesicles from Piscirickettsia salmonis LF-89 type strain identifies plasmid-encoded putative toxins. Front. Cell. Infect. Microbiol. 2017, 7, 420. [Google Scholar] [CrossRef]
- Tandberg, J.I.; Lagos, L.X.; Langlete, P.; Berger, E.; Rishovd, A.L.; Roos, N.; Varkey, D.; Paulsen, I.T.; Winther-Larsen, H.C. Comparative analysis of membrane vesicles from three Piscirickettsia salmonis isolates reveals differences in vesicle characteristics. PLoS ONE 2016, 11, 1–27. [Google Scholar] [CrossRef]
- Yañez, A.J.; Valenzuela, K.; Silva, H.; Retamales, J.; Romero, A.; Enriquez, R.; Figueroa, J.; Claude, A.; Gonzalez, J.; Carcamo, J.G. Broth medium for the successful culture of the fish pathogen Piscirickettsia salmonis. Dis. Aquat. Organ. 2012, 97, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Mandakovic, D.; Glasner, B.; Maldonado, J.; Aravena, P.; Gonzalez, M.; Cambiazo, V.; Pulgar, R. Genomic-based restriction enzyme selection for specific detection of Piscirickettsia salmonis by 16S rDNA PCR-RFLP. Front. Microbiol. 2016, 7, 643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007, 36 (Suppl. 1), D250–D254. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Díaz-Pascual, F.; Ortíz-Severín, J.; Varas, M.A.; Allende, M.L.; Chávez, F.P. In vivo host-pathogen interaction as revealed by global proteomic profiling of zebrafish larvae. Front. Cell. Infect. Microbiol. 2017, 7, 334. [Google Scholar] [CrossRef]
- Varas, M.A.; Riquelme-Barrios, S.; Valenzuela, C.; Marcoleta, A.E.; Berríos-Pastén, C.; Santiviago, C.A.; Chávez, F.P. Inorganic polyphosphate is essential for Salmonella Typhimurium virulence and survival in Dictyostelium discoideum. Front. Cell. Infect. Microbiol. 2018, 8, 8. [Google Scholar] [CrossRef]
- Salazar, C.; Haussmann, D.; Kausel, G.; Figueroa, J. Molecular cloning of Salmo salar Toll-like receptors (TLR1, TLR22, TLR5M and TLR5S) and expression analysis in SHK-1 cells during Piscirickettsia salmonis infection. J. Fish Dis. 2015, 39, 239–248. [Google Scholar] [CrossRef]
- Alvarez, C.A.; Gomez, F.A.; Mercado, L.; Ramirez, R.; Marshall, S.H. Piscirickettsia salmonis imbalances the innate immune response to succeed in a productive infection in a salmonid cell line model. PLoS ONE 2016, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Miranda, D.; Gallardo-Escárate, C. Novel insights into the response of Atlantic salmon (Salmo salar) to Piscirickettsia salmonis: Interplay of coding genes and lncRNAs during bacterial infection. Fish Shellfish Immunol. 2016, 59, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, R.; Gomez, F.A.; Marshall, S.H. The infection process of Piscirickettsia salmonis in fish macrophages is dependent upon interaction with host-cell clathrin and actin. FEMS Microbiol. Lett. 2015, 362, 1–8. [Google Scholar] [CrossRef]
- Dai, S.; Mohapatra, N.P.; Schlesinger, L.S.; Gunn, J.S. Regulation of Francisella tularensis virulence. Front. Microbiol. 2010, 1, 144. [Google Scholar] [CrossRef]
- Cortés, M.P.; Mendoza, S.N.; Travisany, D.; Gaete, A.; Siegel, A.; Cambiazo, V.; Maass, A. Analysis of Piscirickettsia salmonis metabolism using genome-scale reconstruction, modeling, and testing. Front. Microbiol. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Grayfer, L.; Kerimoglu, B.; Yaparla, A.; Hodgkinson, J.W.; Xie, J.; Belosevic, M. Mechanisms of fish macrophage antimicrobial immunity. Front. Immunol. 2018, 9, 1105. [Google Scholar] [CrossRef]
- Jindal, S. Heat shock proteins: Applications in health and disease. Trends Biotechnol. 1996, 14, 17–20. [Google Scholar] [CrossRef]
- Sevin, M.; Girodon, F.; Garrido, C.; De Thonel, A. HSP90 and HSP70: Implication in inflammation processes and therapeutic approaches for myeloproliferative neoplasms. Mediators Inflamm. 2015, 2015, 970242. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 1–12. [Google Scholar]
- Knoops, B.; Argyropoulou, V.; Becker, S.; Ferté, L.; Kuznetsova, O. Multiple roles of peroxiredoxins in inflammation. Mol. Cells 2016, 39, 60–64. [Google Scholar] [PubMed]
- Esteban, M.A.; Chaves-Pozo, E.; Arizcun, M.; Meseguer, J.; Cuesta, A. Regulation of natural killer enhancing factor (NKEF) genes in teleost fish, gilthead seabream and European sea bass. Mol. Immunol. 2013, 55, 275–282. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, Ú.M.; Bron, J.E.; Brown, L.; Pourahmad, F.; Bricknell, I.R.; Thompson, K.D.; Adams, A.; Ellis, A.E. Survival and replication of Piscirickettsia salmonis in rainbow trout head kidney macrophages. Fish Shellfish Immunol. 2008, 25, 477–484. [Google Scholar] [CrossRef]
- Lagos, L.; Tandberg, J.; Kashulin-Bekkelund, A.; Colquhoun, D.; Sørum, H.; Winther-Larsen, H. Isolation and characterization of serum extracellular vesicles (EVs) from Atlantic salmon infected with Piscirickettsia salmonis. Proteomes 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Miranda, D.; Valenzuela-Muñoz, V.; Farlora, R.; Gallardo-Escárate, C. MicroRNA-based transcriptomic responses of Atlantic salmon during infection by the intracellular bacterium Piscirickettsia salmonis. Dev. Comp. Immunol. 2017, 77, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Wei, L.; Cao, Y.; Liu, Q.; Guttman, J.A.; Wang, Q.; Leung, K.Y. Edwardsiella induces microtubule-severing in host epithelial cells. Microbiol. Res. 2019, 229, 126325. [Google Scholar] [CrossRef]
- Alves-Silva, J.; Tavares, I.P.; Guimarães, E.S.; Costa Franco, M.M.; Figueiredo, B.C.; Marques, J.T.; Splitter, G.; Oliveira, S.C. Modulation of microtubule dynamics affects Brucella abortus intracellular survival, pathogen-containing vacuole maturation, and pro-inflammatory cytokine production in infected macrophages. Front. Microbiol. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Radhakrishnan, G.K.; Splitter, G.A. Modulation of host microtubule dynamics by pathogenic bacteria. Biomol. Concepts 2012, 3, 571–580. [Google Scholar] [CrossRef]
- Hendrickson, E.L.; Plotnikova, J.; Mahajan-Miklos, S.; Rahme, L.G.; Ausubel, F.M. Differential roles of the Pseudomonas aeruginosa PA14 rpoN gene in pathogenicity in plants, nematodes, insects, and mice. J. Bacteriol. 2001, 183, 7126–7134. [Google Scholar] [CrossRef]
- Lloyd, M.G.; Lundgren, B.R.; Hall, C.W.; Gagnon, L.B.P.; Mah, T.F.; Moffat, J.F.; Nomura, C.T. Targeting the alternative sigma factor RpoN to combat virulence in Pseudomonas aeruginosa. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Ancona, V.; Li, W.; Zhao, Y. Alternative sigma factor RpoN and its modulation protein YhbH are indispensable for Erwinia amylovora virulence. Mol. Plant Pathol. 2014, 15, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Yu, R.; Schellhorn, H. Antagonistic regulation of motility and transcriptome expression by RpoN and RpoS in Escherichia coli. Mol. Microbiol. 2011, 79, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Gómez, F.A.; Tobar, J.A.; Henríquez, V.; Sola, M.; Altamirano, C.; Marshall, S.H. Evidence of the presence of a functional Dot/Icm type IV-B secretion system in the fish bacterial pathogen Piscirickettsia salmonis. PLoS ONE 2013, 8, e54934. [Google Scholar] [CrossRef]
- Cvitanich, J.D.D.; Garate, N.O.; Smith, C.E.E.; Garate, O.; Smith, C.E.E. The isolation of a rickettsia-like organism causing disease and mortality in Chilean salmonids and its confirmation by Koch’s postulate. J. Fish Dis. 1991, 14, 121–145. [Google Scholar] [CrossRef]
- Mauel, M.J.; Miller, D.L. Piscirickettsiosis and piscirickettsiosis-like infections in fish: A review. Vet. Microbiol. 2002, 87, 279–289. [Google Scholar] [CrossRef]
- Gerlach, R.G.; Hensel, M. Protein secretion systems and adhesins: The molecular armory of Gram-negative pathogens. Int. J. Med. Microbiol. 2007, 297, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Young, G.M.; Schmiel, D.H.; Miller, V.L. A new pathway for the secretion of virulence factors by bacteria: The flagellar export apparatus functions as a protein-secretion system. Proc. Natl. Acad. Sci. USA 1999, 96, 6456–6461. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Sheshachalam, A.; Braverman, N.E.; Rachubinski, R.A.; Simmonds, A.J. Peroxisome-mediated metabolism is required for immune response to microbial infection. Immunity 2017, 47, 93.e7–106.e7. [Google Scholar] [CrossRef]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bülow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in immune response and inflammation. Int. J. Mol. Sci. 2019, 20, 3877. [Google Scholar] [CrossRef]
- Knodler, L.A.; Steele-Mortimer, O. The Salmonella Effector PipB2 affects late endosome/lysosome distribution to mediate sif extension. Mol. Biol. Cell 2005, 16, 4108–4123. [Google Scholar] [CrossRef]
- Ortiz-Severín, J.; Travisany, D.; Maass, A.; Chávez, F.P.; Cambiazo, V. Piscirickettsia salmonis cryptic plasmids: Source of mobile DNA and virulence factors. Pathogens 2019, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, N.F.; Gerloff, D.L.; Uetz, P. Protein domains of unknown function are essential in bacteria. MBio 2014, 5, e00744-13. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; D’haeseleer, P.M.; Branda, S.S.; Liou, M.J.; Haider, Y.; Segelke, B.W.; El-Etr, S.H. Proteomic profiling of Burkholderia thailandensis during host infection using bio-orthogonal noncanonical amino acid tagging (BONCAT). Front. Cell. Infect. Microbiol. 2018, 8, 370. [Google Scholar] [CrossRef]
- Załuga, J.; Stragier, P.; Baeyen, S.; Haegeman, A.; Van Vaerenbergh, J.; Maes, M.; De Vos, P. Comparative genome analysis of pathogenic and non-pathogenic Clavibacter strains reveals adaptations to their lifestyle. BMC Genomics 2014, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UNIPROT ID | Description | Fold-Change Vacuolization vs. Control | Fold-Change Propagation vs. Control | Fold-Change Propagation vs. Vacuolization | Reference |
|---|---|---|---|---|---|
| Iron metabolism | |||||
| B5XCL8 | Lipocalin | V | P | −0.785 | [14] |
| B5X740 | Ferritin | P | P | [9,32] | |
| C0H8L6 | Hemoglobin subunit alpha HBA | P | P | [12] | |
| C0PU56 | Iron-responsive element-binding protein 2 IREB2 | P | P | ||
| Immune response | |||||
| B5X1Q8 | Leukocyte elastase inhibitor ILEU | −0.388 | −0.700 | −0.312 | |
| Q2QEY8 | MHC class I alpha 1 antigen | −4.54 | C | V | |
| B9EPF1 | Proteasome activator complex subunit 1 PSME1 | −3.26 | C | V | |
| B5X3L4 | B-cell lymphoma 6 protein homolog BCL6 | C | −0.994 | P | |
| B9ENT4 | CCAAT/enhancer-binding protein delta | C | 0.580 | P | [7] |
| B9EQ10 | GTPase IMAP family member 7 GIMA7 | C | −0.0038 | P | |
| T2L2I0 | Interleukin-1 receptor-associated kinase 4 | C | −2.05 | P | |
| C0H8Y1 | Interleukin-12 receptor beta-2 chain | C | −1.36 | P | |
| C0H9Y8 | Interleukin-6 receptor subunit alpha | C | −0.321 | P | |
| F5B7Y2 | Membrane toll-like receptor 5 TLR5 | C | −0.321 | P | [12,30] |
| Q8HX42 | MHC class I | C | 0.861 | P | [9] |
| B5X3J8 | Mothers against decapentaplegic homolog SMAD4 | C | −2.23 | P | |
| F5B7Y3 | Soluble toll-like receptor 5 TLR5 | C | −1.02 | P | [12,30] |
| B5X7I0 | TNF receptor-associated factor 5 | C | −1.66 | P | |
| S0F1A6 | Toll-like receptor 8b1 | C | −2.05 | P | |
| B5X6C0 | Gamma-interferon-inducible lysosomal thiol reductase GILT | V | P | −2.42 | [12] |
| B5X1U2 | Interleukin enhancer-binding factor 2 homolog ILF2 | V | P | 4.81 | |
| B5X2T5 | Interleukin enhancer-binding factor 3 homolog ILF3 | V | P | 2.75 | |
| C0PUM0 | CD166 antigen homolog | V | V | ||
| B5X3Y9 | CD99 antigen | V | V | ||
| B5XAB6 | Complement component 1 Q subcomponent-binding protein, mitochondrial | V | V | ||
| B5XFB9 | Complement factor D | V | V | ||
| Q6IWH5 | Interleukin-1 beta | V | V | ||
| C0H963 | Interleukin-17 receptor A | V | V | ||
| C0HAA1 | Interleukin-31 receptor A | V | V | ||
| B9ELT2 | Lysozyme g | V | V | ||
| D0UGE1 | MHC class I antigen Sasa-ZBA | V | V | ||
| B5XAL9 | C-C motif chemokine 13 CCL13 | P | P | [7] | |
| B9ENV6 | CD40 ligand | P | P | ||
| A4ZHU6 | CD4-like protein | P | P | ||
| Q5ILA0 | CD8 beta splice variant | P | P | [7] | |
| C0HBS7 | Complement C1q subcomponent subunit B | P | P | [7] | |
| B5X7J5 | Complement C1q-like protein 2 | P | P | ||
| C0H9W8 | Cytokine receptor-like factor 3 | P | P | ||
| X2JBZ1 | CRFB1b. Type I IFN receptor | P | P | ||
| B5X149 | Interferon regulatory factor 2-binding protein 2-A | P | P | ||
| B5X3F0 | Interferon regulatory factor 2-binding protein 2-B | P | P | ||
| B5X2U0 | Interferon-stimulated 20 kDa exonuclease-like 1 | P | P | ||
| B5XA65 | Lysozyme | P | P | [9] | |
| B9EP53 | Metalloendopeptidase HCE2 | P | P | ||
| C0H9L0 | Metalloendopeptidase BMP1 | P | P | ||
| C0H9S3 | Neutrophil cytosol factor 2 | P | P | ||
| B5X678 | NF-kappa-B inhibitor-interacting Ras-like protein 2 | P | P | ||
| V9Q6E2 | Suppressor of cytokine signaling 2a | P | P | ||
| B9EP15 | T-cell surface antigen CD2 | P | P | ||
| A0A0F6QNL8 | Toll-like receptor 3 | P | P | ||
| B5XCC4 | Tumor necrosis factor receptor superfamily member 11B | P | P | ||
| ROS and stress response | |||||
| B5DG64 | Heat shock protein 90kDa alpha (Cytosolic) class B member 1 | 1.64 | 2.19 | 0.546 | |
| C0HAB6 | Heat shock protein 90-alpha 1 | 1.06 | 1.48 | 0.417 | |
| U5KQM7 | Heat shock protein 90-alpha 2 | 0.883 | 1.48 | 0.598 | [7,8,9] |
| Q9W6K6 | Heat shock protein 90-beta 1 | −0.151 | 1.08 | 1.23 | [7,8,9] |
| B9EMS3 | Heat shock protein 90-beta | 2.89 | 0.733 | −2.16 | [7,8,9] |
| C0HBF1 | 60 kDa heat shock protein CH60, mitochondrial | 2.25 | −2.91 | −5.16 | |
| B5DG30 | Heat shock protein 70 isoform 3 | 0.978 | −0.688 | −1.66 | |
| B5DFX7 | Heat shock cognate 70 kDa protein | 0.187 | −0.906 | −1.09 | |
| B5X3U6 | Heat shock cognate 70 kDa protein HSP70 | −0.0341 | −0.778 | −0.744 | |
| B9ENS1 | Glutathione S-transferase A GSTA | 2.01 | −1.04 | −3.06 | |
| B5X9L4 | Peroxiredoxin-4 PRDX4 | 0.534 | 0.421 | −0.116 | |
| C0HBF4 | Tryparedoxin | 3.31 | −0.503 | −3.81 | |
| B5X5Q6 | Peroxiredoxin-5 PRDX5, mitochondrial | −1.94 | 0.623 | 2.56 | |
| B5X7X7 | Peroxiredoxin-1 PRDX1 | −1.19 | 1.43 | 2.62 | |
| B5X8H5 | Peroxiredoxin TDX | −0.549 | 0.524 | 1.07 | |
| B5X9Q1 | Peroxiredoxin-6 PRDX6 | 2.96 | C | V | |
| B5X0U9 | Dimethylaniline monooxygenase [N-oxide-forming] | 3.20 | C | V | |
| C0PUG5 | Heat shock 70 kDa protein 4 | 1.34 | C | V | |
| Q0KFS6 | Heat shock 90kDa protein | C | 0.777 | P | [7,12] |
| B8YQA0 | NADPH oxidase 1 | C | 0.179 | P | |
| B5XD88 | S-adenosylmethionine synthetase METK2 | C | −3.42 | P | [12] |
| Q8UWF4 | Heat shock protein 60 | C | −4.33 | P | |
| B5DG46 | Stress-induced-phosphoprotein 1 (Hsp70/Hsp90-organizing) stip1 | V | P | −0.577 | |
| U5KQ14 | Heat shock protein 90-alpha 3 | V | P | −0.659 | [7,8,9] |
| B9ELQ1 | Heat shock cognate 71 kDa protein | V | P | −1.01 | |
| B5XGZ2 | Glutathione S-transferase P | V | P | −2.25 | [12] |
| U5KR11 | Heat shock protein 90-alpha 4 | V | P | −2.36 | [7,8,9] |
| B5XDQ7 | Heat shock factor-binding protein 1 HSBP1 | V | V | ||
| Q3ZLR1 | Superoxide dismutase [Cu-Zn] SOD1 | V | V | ||
| Apoptosis | |||||
| B5X8Y9 | Diablo homolog, mitochondrial | C | 0.837 | P | [7] |
| B5XB64 | Thymocyte nuclear protein 1 THYN1 | C | −1.92 | P | |
| B9EL90 | Programmed cell death protein 5 | V | V | ||
| B5DG91 | Apoptosis-associated speck-like protein containing a CARD | V | V | ||
| B5XDE1 | Bcl10-interacting CARD protein | P | P | [7] | |
| B5XEY3 | Caspase-1 | P | P | ||
| B5XDX7 | Programmed cell death 1 ligand 1 PDL1 | P | P | ||
| Cytoskeleton, organization and regulation | |||||
| B5DGE8 | Tubulin alpha chain tuba8l2 | 3.84 | 2.56 | −1.28 | |
| C0HBL4 | Tubulin beta-2A chain | 3.20 | 1.51 | −1.69 | [7] |
| Q2ERI0 | Beta-tubulin | 3.12 | 1.95 | −1.16 | |
| A7KJD9 | TUBB | 3.09 | 1.98 | −1.12 | |
| B5X0U5 | Tubulin beta chain | 2.62 | 1.94 | −0.676 | |
| C0PU76 | Tubulin alpha-1C chain | 1.58 | 1.54 | −0.0359 | |
| B5DH01 | Tubulin alpha chain TBA | 1.55 | 1.80 | 0.251 | |
| B5DH02 | Tubulin, alpha 8 like 3-2 | 1.46 | 1.76 | 0.310 | |
| C0H808 | Tubulin beta-1 chain | 1.42 | 0.766 | −0.650 | |
| B5XFE9 | Tubulin alpha-1A chain | 1.32 | 1.79 | 0.473 | |
| B9EN30 | Tubulin beta-2C chain | 1.28 | 0.714 | −0.568 | |
| B5X3H7 | Tubulin beta-3 chain | 1.27 | −0.500 | −1.77 | |
| B9EMD3 | Tubulin-specific chaperone A | −1.01 | 0.290 | 1.30 | |
| B5XFN3 | Myosin light polypeptide 6B | 1.40 | −0.883 | −2.28 | |
| B5X9N0 | Myosin light polypeptide 6 | 1.30 | −0.837 | −2.14 | |
| C0PU50 | Myosin-9 | 0.562 | 1.77 | 1.21 | |
| B5XAM0 | Thymosin beta-12 | 0.613 | −1.83 | −2.44 | [7,9] |
| B5X2P7 | Tropomyosin-1 alpha chain | −0.300 | −0.118 | 0.182 | |
| B5X2S2 | Tropomyosin alpha-4 chain | −1.11 | 0.0879 | 1.20 | [9] |
| B5X4C0 | Tropomyosin alpha-3 chain | −1.52 | 0.023 | 1.55 | [9] |
| B9ENC8 | Transgelin TAGL | −1.02 | −0.810 | 0.215 | |
| B5X2M3 | Septin-7 | −1.16 | −1.04 | 0.130 | |
| B5XFZ3 | Actin, adductor muscle | 0.546 | 1.51 | 0.962 | |
| B5X3R3 | F-actin-capping protein subunit beta | 0.890 | −1.03 | −1.92 | |
| B5DG56 | Capping protein (actin filament) muscle Z-line α 2 | −0.277 | −1.01 | −0.732 | |
| O42161 | Actin, cytoplasmic 1 | −0.516 | −0.00485 | 0.511 | [9] |
| B5DG40 | Fast myotomal muscle actin 2 | −0.917 | 0.154 | 1.07 | |
| Q78BU2 | Actin alpha 1-1 | −0.967 | 0.104 | 1.07 | |
| C0H9H2 | Actin-related protein 2-A ARP2A | 2.36 | C | V | |
| B5DH12 | Myosin light chain 1-1 | 2.02 | C | V | |
| B5DGD5 | Actin related protein 2/3 complex subunit 2 ARPC2 | 0.179 | C | V | |
| B5XCW2 | Actin-related protein 2/3 complex subunit 4 ARPC4 | C | −2.76 | P | |
| A8WCK1 | Myosin 1 | C | 1.24 | P | |
| C0HBJ2 | Gamma-tubulin complex component 4 | C | −0.519 | P | |
| B5X3R2 | Dynactin subunit 2 | V | P | 1.58 | |
| C0HBE5 | Actin-related protein 3 ARP3 | V | P | −3.50 | |
| C0H8E0 | Actin-related protein 2/3 complex subunit 5 ARPC5 | V | V | ||
| B5RI24 | Actinin alpha 3 | V | V | ||
| C0HBR6 | Actin-related protein 2/3 complex subunit 1B ARC1B | V | V | ||
| C0PUH9 | Actin-related protein 2-B ARP2B | V | V | ||
| B5XFD6 | Myosin light polypeptide 4 | V | V | ||
| B5X1K8 | Myosin regulatory light chain 2, smooth muscle isoform | V | V | ||
| Q7ZZN0 | Myosin regulatory light chain 2 | V | V | ||
| B5XG71 | Actin-related protein 6 ARP6 | P | P | ||
| B5X2S3 | Actin-related protein 8 | P | P | ||
| C0H9Q6 | Rho GTPase-activating protein 15 | P | P | ||
| B5X3B9 | Rho-related GTP-binding protein RhoE | P | P | ||
| Lysosome, phagosome, vesicle fusion and trafficking | |||||
| B5XDV8 | Cystatin-B | 2.69 | −3.43 | −6.12 | [12] |
| B5DFV6 | Cathepsin D | −0.0872 | C | V | [7,31] |
| C0H8J4 | AP-1 complex subunit mu-2 | C | 0.923 | P | [7] |
| B9ELB6 | Charged multivesicular body protein 5 CHMP5 | C | 0.668 | P | |
| C0HBD4 | Interferon-induced guanylate-binding protein 1 GBP1 | C | −0.188 | P | [12,31] |
| C0H9C2 | Phosphatidylinositol-5-phosphate 4-kinase type-2 beta | C | 0.428 | P | |
| B5X6D9 | Ras-related protein ralB-B | C | −0.305 | P | |
| B5X6L9 | Ras-related protein Rab-39B | C | −1.12 | P | |
| B5X127 | Ras-related protein Rab-7a | V | P | 0.251 | |
| C0PUQ5 | Cathepsin Z | V | V | [8] | |
| B5XCD1 | Vesicle-associated membrane protein-associated protein B | V | V | ||
| C0PUP3 | AP-2 complex subunit beta-1 | V | V | [32] | |
| B9EQ28 | Clathrin light chain B | V | V | [32] | |
| C0HAE8 | Phosphatidylinositol-binding clathrin assembly protein PICA | V | V | ||
| C0HAE8 | Phosphatidylinositol-binding clathrin assembly protein | V | V | ||
| B9ENL7 | Ras-related protein Rab-5C | V | V | ||
| B5XFU5 | Adaptin ear-binding coat-associated protein 1 | P | P | ||
| B5X152 | AP-3 complex subunit mu-1 | P | P | ||
| B5RI38 | Cathepsin l-like | P | P | [12] | |
| C0HA36 | Endophilin-B2 | P | P | ||
| C0H980 | Lysosome-associated membrane glycoprotein 1 LAMP1 | P | P | ||
| B5X7X4 | Ragulator complex protein LAMTOR2 | P | P | ||
| B5X198 | Ran GTPase-activating protein 1 RGP1 | P | P | ||
| C0PU44 | Ras-related protein Rab-14 | P | P | ||
| B5X0U0 | Ras-related protein Rab-18 | P | P | ||
| B5X481 | Synaptotagmin-4 SYT4 | P | P | ||
| B5X179 | Syntaxin-3 | P | P | ||
| C0H9V3 | Syntaxin-5 | P | P | ||
| C0PUM9 | Syntaxin-binding protein 2 STXB2 | P | P | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Severín, J.; Travisany, D.; Maass, A.; Cambiazo, V.; Chávez, F.P. Global Proteomic Profiling of Piscirickettsia salmonis and Salmon Macrophage-Like Cells during Intracellular Infection. Microorganisms 2020, 8, 1845. https://doi.org/10.3390/microorganisms8121845
Ortiz-Severín J, Travisany D, Maass A, Cambiazo V, Chávez FP. Global Proteomic Profiling of Piscirickettsia salmonis and Salmon Macrophage-Like Cells during Intracellular Infection. Microorganisms. 2020; 8(12):1845. https://doi.org/10.3390/microorganisms8121845
Chicago/Turabian StyleOrtiz-Severín, Javiera, Dante Travisany, Alejandro Maass, Verónica Cambiazo, and Francisco P. Chávez. 2020. "Global Proteomic Profiling of Piscirickettsia salmonis and Salmon Macrophage-Like Cells during Intracellular Infection" Microorganisms 8, no. 12: 1845. https://doi.org/10.3390/microorganisms8121845
APA StyleOrtiz-Severín, J., Travisany, D., Maass, A., Cambiazo, V., & Chávez, F. P. (2020). Global Proteomic Profiling of Piscirickettsia salmonis and Salmon Macrophage-Like Cells during Intracellular Infection. Microorganisms, 8(12), 1845. https://doi.org/10.3390/microorganisms8121845