The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Physicochemical Characterization
2.2. Sample Collection and Processing from Chignahuapan Puebla, Mexico
2.3. Taxonomic Annotation of Metagenome
2.4. Co-Occurrence Network Analysis
2.5. Identification and Annotation of Viral Genomes
2.6. Functional Annotation
3. Results and Discussion
3.1. Field Sampling and Physicochemical Characterization
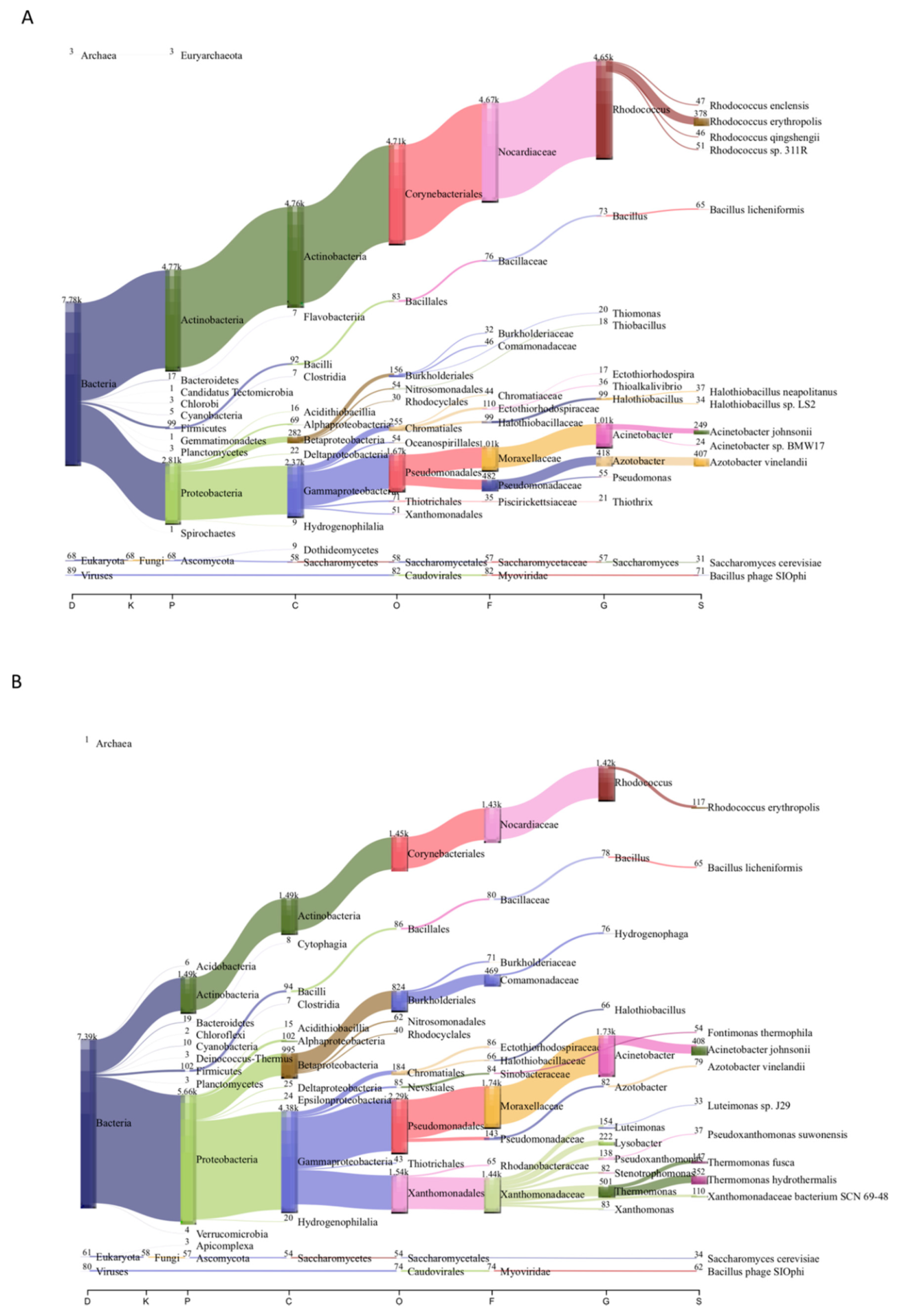
3.2. Microbial Community Composition of Chignahuapan Metagenomes
3.3. Comparative Analysis and Ecological Indices
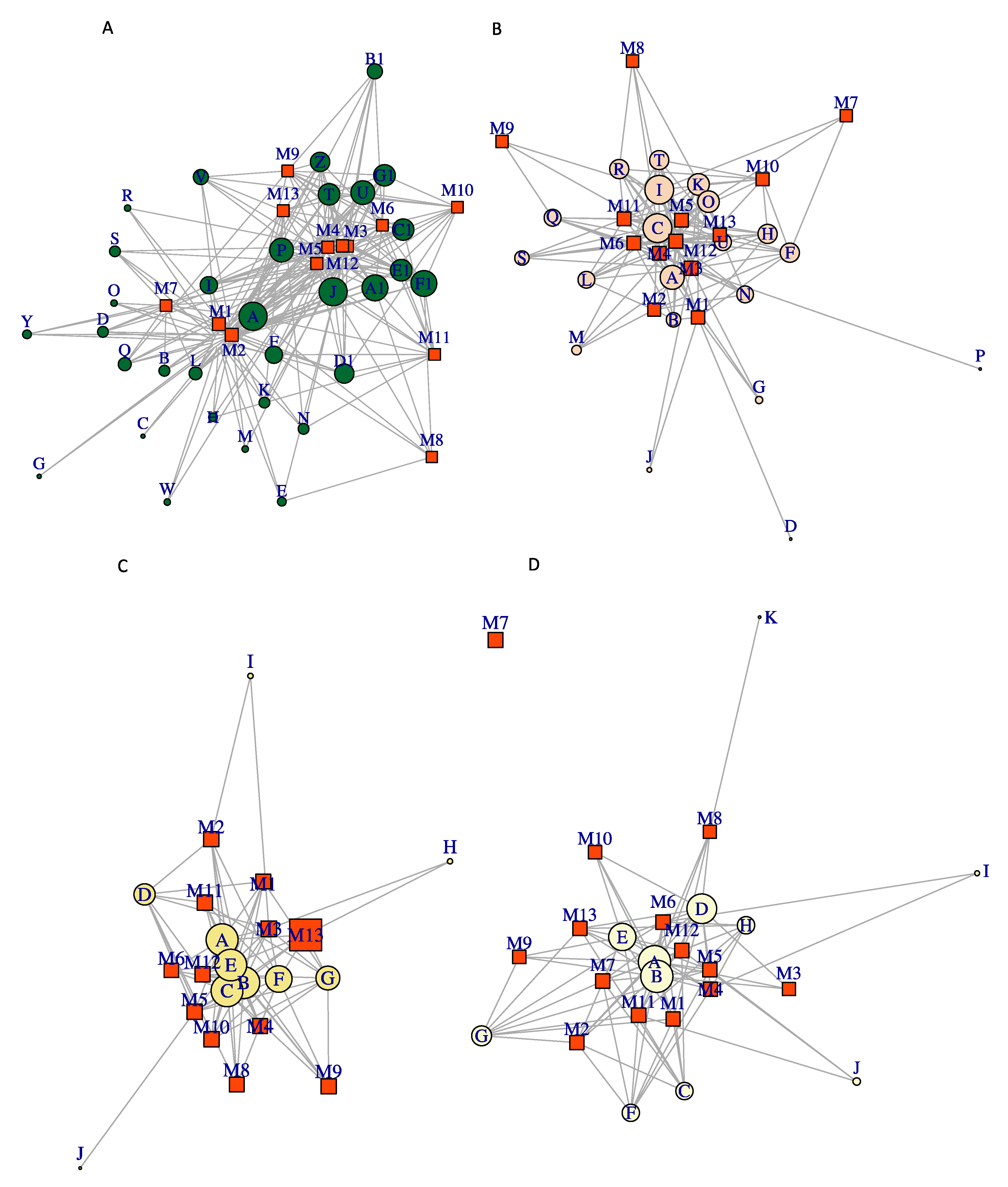
3.4. Co-Occurrence Network Analysis
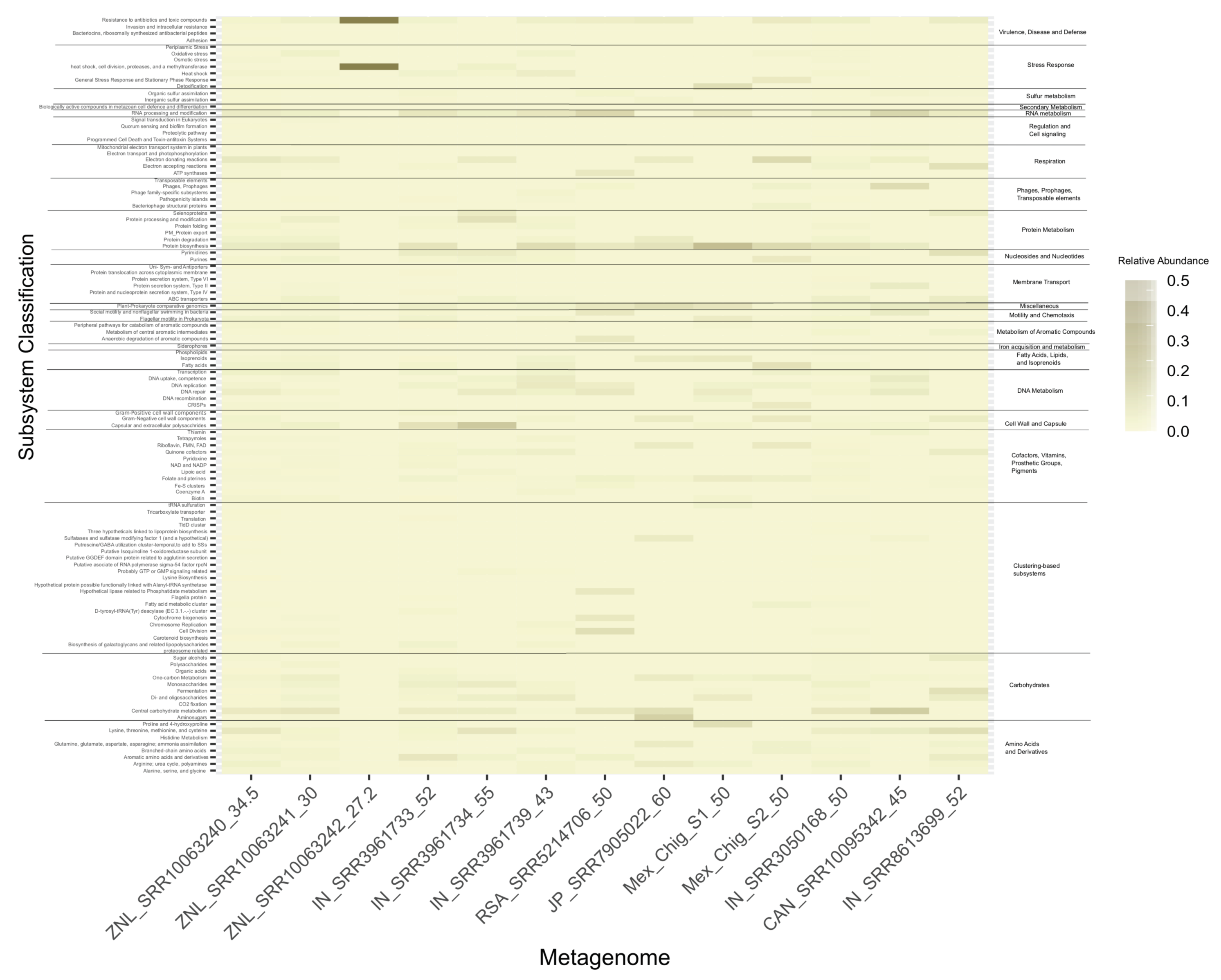
3.5. Functional Metagenomics Analysis
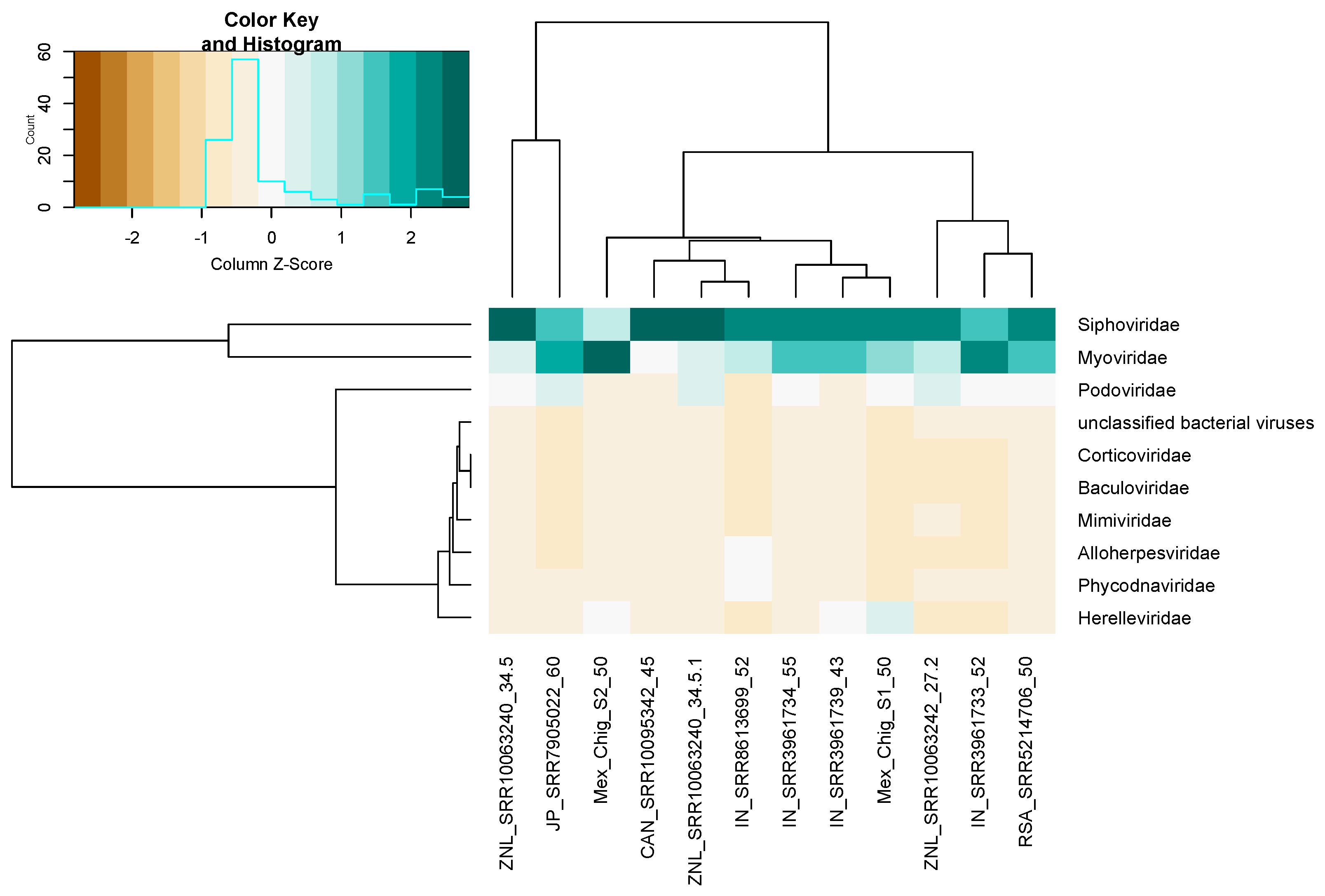
3.6. Viral Community Composition
3.7. Auxiliary Metabolic Genes and Whole Viral Genomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Inskeep, W.P.; Jay, Z.J.; Tringe, S.G.; Herrgard, M.; Rusch, D.B. The YNP Metagenome project: Environmental parameters responsible for microbial distribution in the yellowstone geothermal ecosystem. Front. Microbiol. 2013, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Mardanov, A.V.; Gumerov, V.M.; Beletsky, A.V.; Ravin, N.V. Microbial diversity in acidic thermal pools in the Uzon Caldera, Kamchatka. Antonie Van Leeuwenhoek 2018, 111, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Strazzulli, A.; Fusco, S.; Cobucci-Ponzano, B.; Moracci, M.; Contursi, P. Metagenomics of microbial and viral life in terrestrial geothermal environments. Rev. Environ. Sci. Biotechnol. 2017, 16, 425–454. [Google Scholar] [CrossRef]
- Arsanova, G.I. The origin of thermal waters in volcanic areas. J. Volcanol. Seismol. 2014, 8, 361–374. [Google Scholar] [CrossRef]
- Inskeep, W.P.; Jay, Z.J.; Herrgard, M.J.; Kozubal, M.A.; Rusch, D.B.; Tringe, S.G.; Macur, R.E.; deM. Jennings, R.; Boyd, E.S.; Spear, J.R.; et al. Phylogenetic and functional analysis of metagenome sequence from high-temperature archaeal habitats demonstrate linkages between metabolic potential and geochemistry. Front. Microbiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Valeriani, F.; Crognale, S.; Protano, C.; Gianfranceschi, G.; Orsini, M.; Vitali, M.; Spica, V.R. Metagenomic analysis of bacterial community in a travertine depositing hot spring. New Microbiol. 2018, 41, 126–135. [Google Scholar]
- López-López, O.; Cerdán, M.E.; González-Siso, M.I. Hot spring metagenomics. Life 2013, 3, 308–320. [Google Scholar] [CrossRef]
- Marsh, C.L.; Larsen, D.H. CHARACTERIZATION of some thermophilic bacteria from the hot springs of yellowstone national park. J. Bacteriol. 1953, 65, 193–197. [Google Scholar] [CrossRef]
- Saxena, R.; Dhakan, D.B.; Mittal, P.; Waiker, P.; Chowdhury, A.; Ghatak, A.; Sharma, V.K. Metagenomic analysis of hot springs in central india reveals hydrocarbon degrading thermophiles and pathways essential for survival in extreme environments. Front. Microbiol. 2017, 7, 2123. [Google Scholar] [CrossRef]
- Mangrola, A.; Dudhagara, P.; Koringa, P.; Joshi, C.G.; Parmar, M.; Patel, R. Deciphering the microbiota of Tuwa hot spring, India using shotgun metagenomic sequencing approach. Genom. Data 2015, 4, 153–155. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Subudhi, E.; Kumar, M. Investigation of bacterial diversity of hot springs of Odisha, India. Genom. Data 2015, 6, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, N.; Lambert, C.; Sharma, A.; Gupta, V.; Khurana, P.; Khurana, J.P.; Sockett, R.E.; Gilbert, J.A.; Lal, R. Arsenic rich Himalayan hot spring metagenomics reveal genetically novel predator-prey genotypes: Metagenomic recovery of predator prey genotypes. Environ. Microbiol. Rep. 2015, 7, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Ahmed, I.; Salam, N.; Kim, B.-Y.; Singh, D.; Zhi, X.-Y.; Xiao, M.; Li, W.-J. Diversity and distribution of thermophilic bacteria in hot springs of pakistan. Microb. Ecol. 2017, 74, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.K.; Bisht, S.S.; De Mandal, S.; Kumar, N.S. Bacterial and archeal community composition in hot springs from Indo-Burma region, North-East India. AMB Express 2016, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.J.; Andreote, F.D.; Chaves, D.; Montaña, J.S.; Osorio-Forero, C.; Junca, H.; Zambrano, M.M.; Baena, S. Structural and functional insights from the metagenome of an acidic hot spring microbial Planktonic Community in the Colombian Andes. PLoS ONE 2012, 7, e52069. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Cortez, Y.; Vera, N.; Villena, G.K.; Gutiérrez-Correa, M. Metagenomic analysis of microbial community of an Amazonian geothermal spring in Peru. Genom. Data 2016, 9, 63–66. [Google Scholar] [CrossRef]
- Hussein, E.I.; Jacob, J.H.; Shakhatreh, M.A.K.; Al-razaq, M.A.A.; Juhmani, A.F.; Cornelison, C.T. Exploring the microbial diversity in Jordanian hot springs by comparative metagenomic analysis. MicrobiologyOpen 2017, 6, e00521. [Google Scholar] [CrossRef]
- Kubo, K.; Knittel, K.; Amann, R.; Fukui, M.; Matsuura, K. Sulfur-metabolizing bacterial populations in microbial mats of the Nakabusa hot spring, Japan. Syst. Appl. Microbiol. 2011, 34, 293–302. [Google Scholar] [CrossRef]
- Nishiyama, E.; Higashi, K.; Mori, H.; Suda, K.; Nakamura, H.; Omori, S.; Maruyama, S.; Hongoh, Y.; Kurokawa, K. The relationship between Microbial Community Structures and Environmental Parameters Revealed by Metagenomic Analysis of hot spring water in the Kirishima Area, Japan. Front. Bioeng. Biotechnol. 2018, 6, 202. [Google Scholar] [CrossRef]
- Tobler, D.J.; Benning, L.G. Bacterial diversity in five Icelandic geothermal waters: Temperature and sinter growth rate effects. Extremophiles 2011, 15, 473. [Google Scholar] [CrossRef]
- Chan, C.S.; Chan, K.-G.; Ee, R.; Hong, K.-W.; Urbieta, M.S.; Donati, E.R.; Shamsir, M.S.; Goh, K.M. Effects of physiochemical factors on prokaryotic biodiversity in malaysian circumneutral hot springs. Front. Microbiol. 2017, 8, 1252. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Gudbergsdóttir, S.R.; Rike, A.G.; Lin, L.; Zhang, Q.; Contursi, P.; Moracci, M.; Kristjansson, J.K.; Bolduc, B.; Gavrilov, S.; et al. Comparative metagenomics of eight geographically remote terrestrial hot springs. Microb. Ecol. 2015, 70, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Inskeep, W.P.; Rusch, D.B.; Jay, Z.J.; Herrgard, M.J.; Kozubal, M.A.; Richardson, T.H.; Macur, R.E.; Hamamura, N.; deM. Jennings, R.; Fouke, B.W.; et al. Metagenomes from High-Temperature Chemotrophic Systems Reveal Geochemical Controls on Microbial Community Structure and Function. PLoS ONE 2010, 5, e9773. [Google Scholar] [CrossRef] [PubMed]
- López-López, O.; Knapik, K.; Cerdán, M.-E.; González-Siso, M.-I. Metagenomics of an alkaline hot spring in Galicia (Spain): Microbial diversity analysis and screening for novel lipolytic enzymes. Front. Microbiol. 2015, 6, 1291. [Google Scholar] [CrossRef]
- Brito, E.M.S.; Villegas-Negrete, N.; Sotelo-González, I.A.; Caretta, C.A.; Goñi-Urriza, M.; Gassie, C.; Hakil, F.; Colin, Y.; Duran, R.; Gutiérrez-Corona, F.; et al. Microbial diversity in Los Azufres geothermal field (Michoacán, Mexico) and isolation of representative sulfate and sulfur reducers. Extremophiles 2014, 18, 385–398. [Google Scholar] [CrossRef]
- Massello, F.L.; Chan, C.S.; Chan, K.-G.; Goh, K.M.; Donati, E.; Urbieta, M.S. Meta-Analysis of microbial communities in hot springs: Recurrent taxa and complex shaping factors beyond PH and temperature. Microorganisms 2020, 8, 906. [Google Scholar] [CrossRef]
- Sánchez-Córdova, M.M.; Canet, C.; Rodríguez-Díaz, A.; González-Partida, E.; Linares-López, C. Water-rock interactions in the Acoculco geothermal system, eastern Mexico: Insights from paragenesis and elemental mass-balance. Geochemistry 2020, 80, 125527. [Google Scholar] [CrossRef]
- López-Hernández, A.; García-Estrada, G.; Aguirre-Díaz, G.; González-Partida, E.; Palma-Guzmán, H.; Quijano-León, J.L. Hydrothermal activity in the Tulancingo–Acoculco Caldera Complex, central Mexico: Exploratory studies. Geothermics 2009, 38, 279–293. [Google Scholar] [CrossRef]
- Servín-Garcidueñas, L.E.; Martínez-Romero, E. Draft genome sequence of the Sulfolobales Archaeon AZ1, obtained through metagenomic analysis of a Mexican Hot Spring. Genome Announc. 2014, 2, e00164-14. [Google Scholar] [CrossRef]
- Servin-Garciduenas, L.E.; Peng, X.; Garrett, R.A.; Martinez-Romero, E. Genome sequence of a novel archaeal rudivirus recovered from a Mexican Hot Spring. Genome Announc. 2013, 1, e00040-12. [Google Scholar] [CrossRef]
- Prieto-Barajas, C.M.; Alcaraz, L.D.; Valencia-Cantero, E.; Santoyo, G. Life in hot spring microbial mats located in the Trans-Mexican Volcanic Belt: A 16S/18S rRNA gene and metagenomic analysis. Geomicrobiol. J. 2018, 35, 704–712. [Google Scholar] [CrossRef]
- Prieto-Barajas, C.M.; Alfaro-Cuevas, R.; Valencia-Cantero, E.; Santoyo, G. Effect of seasonality and physicochemical parameters on bacterial communities in two hot spring microbial mats from Araró, Mexico. Rev. Mex. Biodivers. 2017, 88, 616–624. [Google Scholar] [CrossRef]
- Pinzón-Martínez, D.L.; Rodríguez-Gómez, C.; Miñana-Galbis, D.; Carrillo-Chávez, J.A.; Valerio-Alfaro, G.; Oliart-Ros, R. Thermophilic bacteria from Mexican thermal environments: Isolation and potential applications. Environ. Technol. 2010, 31, 957–966. [Google Scholar] [CrossRef] [PubMed]
- NMX-AA-008-SCFI-2016: ANÁLISIS DE AGUA.—MEDICIÓN DEL pH EN AGUAS NATURALES, RESIDUALES Y RESIDUALES TRATADAS.-MÉTODO DE PRUEBA-(CANCELA A LA NMX-AA-008SCFI-2011). Available online: https://www.gob.mx/cms/uploads/attachment/file/166767/NMX-AA-008-SCFI-2016.pdf (accessed on 21 August 2019).
- NMX-AA-079-SCFI-2001: ANÁLISIS DE AGUAS—DETERMINACIÓN DE NITRATOS EN AGUAS NATURALES, POTABLES, RESIDUALES Y RESIDUALES TRATADAS—MÉTODO DE PRUEBA (CANCELA A LA NMX-AA-079-1986). Available online: http://biblioteca.semarnat.gob.mx/janium/Documentos/Ciga/agenda/PPD1/DO88.pdf (accessed on 21 August 2019).
- NMX-AA-074-SCFI-2014: ANÁLISIS DE AGUA—MEDICIÓN DEL ION SULFATO EN AGUAS NATURALES, RESIDUALES Y RESIDUALES TRATADAS—MÉTODO DE PRUEBA—(CANCELA A LA NMX-AA-074-1981). Available online: http://201.116.60.182/CONAGUA07/Noticias/NMX-AA-074-2014.pdf (accessed on 21 August 2019).
- NMX-AA-029-SCFI-2001: ANÁLISIS DE AGUAS—DETERMINACIÓN DE FÓSFORO TOTAL EN AGUAS NATURALES, RESIDUALES Y RESIDUALES TRATADAS—MÉTODO DE PRUEBA (CANCELA A LA NMX-AA029-1981). Available online: https://www.gob.mx/cms/uploads/attachment/file/166773/NMX-AA-029-SCFI-2001.pdf (accessed on 21 August 2019).
- NMX-AA-073-SCFI-2001: ANÁLISIS DE AGUA—DETERMINACIÓN DE CLORUROS TOTALES EN AGUAS NATURALES, RESIDUALES Y RESIDUALES TRATADAS—MÉTODO DE PRUEBA (CANCELA A LA NMX-AA-073-1981). Available online: https://www.gob.mx/cms/uploads/attachment/file/166789/NMX-AA-073-SCFI-2001.pdf (accessed on 21 August 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 2019, btz715. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 10 June 2020).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Team, T.I.C. Igraph; Zenodo: Meyrin, Switzerland, 2020. [Google Scholar]
- Dormann, C.F.; Frund, J.; Bluthgen, N.; Gruber, B. Indices, graphs and null models: Analyzing bipartite ecological networks. Open Ecol. J. 2009, 2, 7–24. [Google Scholar] [CrossRef]
- Alcalá-Briseño, R.I.; Casarrubias-Castillo, K.; López-Ley, D.; Garrett, K.A.; Silva-Rosales, L. Network analysis of the papaya orchard virome from two agroecological regions of Chiapas, Mexico. mSystems 2020, 5, e00423-19. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Kawai, S. An algorithm for drawing general undirected graphs. Inf. Process. Lett. 1989, 31, 7–15. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Virome Function from Genomic Sequences|bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/855387v1 (accessed on 10 June 2020).
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, Use R! 2nd ed.; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. In Microbial Environmental Genomics (MEG); Martin, F., Uroz, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 207–233. ISBN 978-1-4939-3369-3. [Google Scholar]
- Wang, J.; Yang, D.; Zhang, Y.; Shen, J.; van der Gast, C.; Hahn, M.W.; Wu, Q. Do Patterns of bacterial diversity along salinity gradients differ from those observed for macroorganisms? PLoS ONE 2011, 6, e27597. [Google Scholar] [CrossRef]
- Castelán-Sánchez, H.G.; Elorrieta, P.; Romoacca, P.; Liñan-Torres, A.; Sierra, J.L.; Vera, I.; Batista-García, R.A.; Tenorio-Salgado, S.; Lizama-Uc, G.; Pérez-Rueda, E.; et al. Intermediate-Salinity systems at high altitudes in the peruvian andes unveil a high diversity and abundance of bacteria and viruses. Genes 2019, 10, 891. [Google Scholar] [CrossRef]
- Ohhata, N.; Yoshida, N.; Egami, H.; Katsuragi, T.; Tani, Y.; Takagi, H. An extremely oligotrophic bacterium, rhodococcus erythropolis N9T-4, isolated from crude oil. J. Bacteriol. 2007, 189, 6824–6831. [Google Scholar] [CrossRef]
- Castorena, G.; Suárez, C.; Valdez, I.; Amador, G.; Fernández, L.; Le Borgne, S. Sulfur-selective desulfurization of dibenzothiophene and diesel oil by newly isolated Rhodococcus sp. strains. FEMS Microbiol. Lett. 2002, 215, 157–161. [Google Scholar] [CrossRef]
- Veith, A.; Botelho, H.M.; Kindinger, F.; Gomes, C.M.; Kletzin, A. The sulfur oxygenase reductase from the mesophilic bacterium halothiobacillus neapolitanus is a highly active thermozyme. J. Bacteriol. 2012, 194, 677–685. [Google Scholar] [CrossRef]
- Mohammad, B.T.; Al Daghistani, H.I.; Jaouani, A.; Abdel-Latif, S.; Kennes, C. Isolation and Characterization of Thermophilic Bacteria from Jordanian Hot Springs: Bacillus Licheniformis and Thermomonas Hydrothermalis Isolates as Potential Producers of Thermostable Enzymes. Available online: https://www.hindawi.com/journals/ijmicro/2017/6943952/ (accessed on 16 June 2020).
- Rey, M.W.; Ramaiya, P.; Nelson, B.A.; Brody-Karpin, S.D.; Zaretsky, E.J.; Tang, M.; de Leon, A.L.; Xiang, H.; Gusti, V.; Clausen, I.G.; et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 2004, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef]
- Vieille, C.; Zeikus, G.J. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001, 65, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Jyoti, V.; Bhadra, B.; Nayak, K.C.; Shivaji, S.; Rainey, F.A.; Das, S.K. Thiomonas bhubaneswarensis sp. nov., an obligately mixotrophic, moderately thermophilic, thiosulfate-oxidizing bacterium. Int. J. Syst. Evol. Microbiol. 2009, 59, 2171–2175. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Chan, K.-G.; Tay, Y.-L.; Chua, Y.-H.; Goh, K.M. Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front. Microbiol. 2015, 6, 177. [Google Scholar] [CrossRef]
- Kvist, T.; Mengewein, A.; Manzei, S.; Ahring, B.K.; Westermann, P. Diversity of thermophilic and non-thermophilic crenarchaeota at 80 °C. FEMS Microbiol. Lett. 2005, 244, 61–68. [Google Scholar] [CrossRef]
- Probst, A.J.; Weinmaier, T.; Raymann, K.; Perras, A.; Emerson, J.B.; Rattei, T.; Wanner, G.; Klingl, A.; Berg, I.A.; Yoshinaga, M.; et al. Biology of a widespread uncultivated archaeon that contributes to carbon fixation in the subsurface. Nat. Commun. 2014, 5, 5497. [Google Scholar] [CrossRef]
- Baker, B.J.; De Anda, V.; Seitz, K.W.; Dombrowski, N.; Santoro, A.E.; Lloyd, K.G. Diversity, ecology and evolution of Archaea. Nat. Microbiol. 2020, 5, 887–900. [Google Scholar] [CrossRef]
- Mehetre, G.T.; Paranjpe, A.S.; Dastager, S.G.; Dharne, M.S. Complete metagenome sequencing based bacterial diversity and functional insights from basaltic hot spring of Unkeshwar, Maharashtra, India. Genom. Data 2016, 7, 140–143. [Google Scholar] [CrossRef]
- Stewart, L.C.; Stucker, V.K.; Stott, M.B.; de Ronde, C.E.J. Marine-influenced microbial communities inhabit terrestrial hot springs on a remote island volcano. Extremophiles 2018, 22, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.M.; Idei, A.; Nakagawa, M.; Ueno, Y.; Fischer, W.W.; McGlynn, S.E. Geochemical and metagenomic characterization of jinata onsen, a proterozoic-analog hot spring, reveals novel microbial diversity including iron-tolerant phototrophs and thermophilic lithotrophs. Microbes Environ. 2019, 34, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.E.; Brady, A.L.; Sharp, G.H.; Grasby, S.E.; Stott, M.B.; Dunfield, P.F. Humboldt’s spa: Microbial diversity is controlled by temperature in geothermal environments. ISME J. 2014, 8, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Hügler, M.; Ward, D.M.; Bryant, D.A. The dark side of the mushroom spring microbial mat: Life in the Shadow of Chlorophototrophs. II. Metabolic Functions of abundant community members predicted from metagenomic analyses. Front. Microbiol. 2017, 8, 943. [Google Scholar] [CrossRef] [PubMed]
- Nunoura, T.; Chikaraishi, Y.; Izaki, R.; Suwa, T.; Sato, T.; Harada, T.; Mori, K.; Kato, Y.; Miyazaki, M.; Shimamura, S.; et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 2018, 359, 559–563. [Google Scholar] [CrossRef]
- Gai, C.S.; Lu, J.; Brigham, C.J.; Bernardi, A.C.; Sinskey, A.J. Insights into bacterial CO2 metabolism revealed by the characterization of four carbonic anhydrases in Ralstonia eutropha H16. AMB Express 2014, 4, 2. [Google Scholar] [CrossRef]
- Bryan, C.G.; Davis-Belmar, C.S.; van Wyk, N.; Fraser, M.K.; Dew, D.; Rautenbach, G.F.; Harrison, S.T.L. The effect of CO2 availability on the growth, iron oxidation and CO2-fixation rates of pure cultures of Leptospirillum ferriphilum and Acidithiobacillus ferrooxidans. Biotechnol. Bioeng. 2012, 109, 1693–1703. [Google Scholar] [CrossRef]
- Schenk, A.; Aragno, M. Bacillus schlegelii, a New species of thermophilic, facultatively chemolithoautotrophic bacterium oxidizing molecular hydrogen. J. Gen. Microbiol. 1979, 115, 333–341. [Google Scholar] [CrossRef]
- Hamana, K.; Matsuzaki, S. Polyamines of carbon monoxide-utilizing bacteria, Pseudomonas thermocarboxydovorans and Pseudomonas carboxydohydrogena. FEMS Microbiol. Lett. 1990, 70, 353–356. [Google Scholar] [CrossRef][Green Version]
- Stipanuk, M.H.; Ueki, I. Dealing with methionine/homocysteine sulfur: Cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011, 34, 17–32. [Google Scholar] [CrossRef]
- Ruby, E.G.; Jannasch, H.W. Chemolithotrophic sulfur-oxidizing bacteria from the galapagos rift hydrothermal ventst. Appl. Environ. Microbiol. 1981, 42, 8. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, G.; Artacho, A.; Rojas, R.A.; Bautista, J.S.; Méndez, R.; Gamboa, M.T.; Gamboa, J.R.; Gómez-Cruz, R. Metagenomics of bacterial diversity in villa luz caves with sulfur water springs. Genes 2018, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Denome, S.A.; Oldfield, C.; Nash, L.J.; Young, K.D. Characterization of the desulfurization genes from Rhodococcus sp. strain IGTS8. J. Bacteriol. 1994, 176, 6707–6716. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, P.; Chen, Z.; Li, Y.; Li, Y.; Wang, Y.; Zhou, J.; Li, G.; Ma, T. Compositions and abundances of sulfate-reducing and sulfur-oxidizing microorganisms in water-flooded petroleum reservoirs with different temperatures in China. Front. Microbiol. 2017, 8, 143. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takai, K. Sulfur metabolisms in epsilon- and gamma-proteobacteria in deep-sea hydrothermal fields. Front. Microbiol. 2011, 2, 192. [Google Scholar] [CrossRef]
- Fishbain, S.; Dillon, J.G.; Gough, H.L.; Stahl, D.A. Linkage of high rates of sulfate reduction in Yellowstone Hot Springs to unique sequence types in the dissimilatory sulfate respiration pathway. Appl. Environ. Microbiol. 2003, 69, 3663–3667. [Google Scholar] [CrossRef] [PubMed]
- Dillon, J.G.; Fishbain, S.; Miller, S.R.; Bebout, B.M.; Habicht, K.S.; Webb, S.M.; Stahl, D.A. High rates of sulfate reduction in a Low-Sulfate Hot Spring Microbial Mat are driven by a low level of diversity of sulfate-respiring microorganisms. Appl. Environ. Microbiol. 2007, 73, 5218–5226. [Google Scholar] [CrossRef]
- Nishihara, A.; Haruta, S.; McGlynn, S.E.; Thiel, V.; Matsuura, K. Nitrogen fixation in thermophilic chemosynthetic microbial communities depending on hydrogen, sulfate, and carbon dioxide. Microbes Environ. 2018, 33, 10–18. [Google Scholar] [CrossRef]
- Ranawat, P.; Rawat, S. Stress response physiology of thermophiles. Arch. Microbiol. 2017, 199, 391–414. [Google Scholar] [CrossRef]
- Dávila-Ramos, S.; Castelán-Sánchez, H.G.; Martínez-Ávila, L.; Sánchez-Carbente, M.d.R.; Peralta, R.; Hernández-Mendoza, A.; Dobson, A.D.W.; Gonzalez, R.A.; Pastor, N.; Batista-García, R.A. A Review on viral metagenomics in extreme environments. Front. Microbiol. 2019, 10, 2403. [Google Scholar] [CrossRef]
- Snyder, J.C.; Stedman, K.; Rice, G.; Wiedenheft, B.; Spuhler, J.; Young, M.J. Viruses of hyperthermophilic Archaea. Res. Microbiol. 2003, 154, 474–482. [Google Scholar] [CrossRef]
- Pina, M.; Bize, A.; Forterre, P.; Prangishvili, D. The archeoviruses. FEMS Microbiol. Rev. 2011, 35, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Barylski, J.; Kropinski, A.M.; Alikhan, N.-F.; Adriaenssens, E.M. ICTV report consortium ICTV virus taxonomy profile: Herelleviridae. J. Gen. Virol. 2020, 101, 362–363. [Google Scholar] [CrossRef] [PubMed]
- Mangold, S.; Valdés, J.; Holmes, D.; Dopson, M. Sulfur metabolism in the extreme acidophile acidithiobacillus caldus. Front. Microbiol. 2011, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Castelán-Sánchez, H.G.; Lopéz-Rosas, I.; García-Suastegui, W.A.; Peralta, R.; Dobson, A.D.W.; Batista-García, R.A.; Dávila-Ramos, S. Extremophile deep-sea viral communities from hydrothermal vents: Structural and functional analysis. Mar. Genom. 2019, 46, 16–28. [Google Scholar] [CrossRef]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B.H. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Li, H.; Zhang, X. Deep-Sea hydrothermal vent viruses compensate for microbial metabolism in virus-host interactions. mBio 2017, 8, 00893-17. [Google Scholar] [CrossRef]
- Puxty, R.J.; Evans, D.J.; Millard, A.D.; Scanlan, D.J. Energy limitation of cyanophage development: Implications for marine carbon cycling. ISME J. 2018, 12, 1273–1286. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef]
- White, R.H. Distribution of folates and modified folates in extremely thermophilic bacteria. J. Bacteriol. 1991, 173, 1987–1991. [Google Scholar] [CrossRef]
- Hutinet, G.; Kot, W.; Cui, L.; Hillebrand, R.; Balamkundu, S.; Gnanakalai, S.; Neelakandan, R.; Carstens, A.B.; Fa Lui, C.; Tremblay, D.; et al. 7-Deazaguanine modifications protect phage DNA from host restriction systems. Nat. Commun. 2019, 10, 5442. [Google Scholar] [CrossRef] [PubMed]
- Pannekens, M.; Kroll, L.; Müller, H.; Mbow, F.T.; Meckenstock, R.U. Oil reservoirs, an exceptional habitat for microorganisms. New Biotechnol. 2019, 49, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Duhaime, M.B.; Breier, J.A.; Wendt, K.A.; Toner, B.M.; Dick, G.J. Sulfur oxidation genes in diverse deep-sea viruses. Science 2014, 344, 757–760. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Properties | Mex_Chig_S1 | Mex_Chig_S2 |
|---|---|---|
| Temperature in °C | 49–50 | 45 |
| pH | 7.02 | 6.66 |
| Electrical conductivity dS m−1 | 1.52 | 1.50 |
| Ca2+ mg L−1 | 203.1 | 81.6 |
| Mg2+ mg L−1 | 33.2 | 17.3 |
| Na1+ mg L−1 | 102.0 | 212.9 |
| K1+ mg L−1 | 14.6 | 12.8 |
| NO3−1 mg L−1 | 16.9 | 12.2 |
| SO4−2 mg L−1 | 25.6 | 30.2 |
| PO4−3 mg L−1 | 4.2 | 2.5 |
| CO3−2 mg L−1 | 0 | 0 |
| HCO3−1 mg L−1 | 780.8 | 634.4 |
| Cl−1 mg L−1 | 196.0 | 98.9 |
| Sample | No. Total of Contigs | Total Contigs | % Contigs Classified within Domain | ||||
|---|---|---|---|---|---|---|---|
| Classified | Unclassified | Bacteria | Archaea | Eukarya | Virus | ||
| Mex_Chig_S1 | 8474 | 8082 (95.5%) | 392 (4.51%) | 91.8% | 1.8% | 0.802% | 1.08% |
| Mex_Chig_S2 | 8361 | 7645 (91.4%) | 716 (8.56%) | 88.4% | 1.3% | 0.7299% | 0.957% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelán-Sánchez, H.G.; Meza-Rodríguez, P.M.; Carrillo, E.; Ríos-Vázquez, D.I.; Liñan-Torres, A.; Batista-García, R.A.; Pérez-Rueda, E.; Rojas-Ruíz, N.E.; Dávila-Ramos, S. The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities. Microorganisms 2020, 8, 1677. https://doi.org/10.3390/microorganisms8111677
Castelán-Sánchez HG, Meza-Rodríguez PM, Carrillo E, Ríos-Vázquez DI, Liñan-Torres A, Batista-García RA, Pérez-Rueda E, Rojas-Ruíz NE, Dávila-Ramos S. The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities. Microorganisms. 2020; 8(11):1677. https://doi.org/10.3390/microorganisms8111677
Chicago/Turabian StyleCastelán-Sánchez, Hugo Gildardo, Pablo M. Meza-Rodríguez, Erika Carrillo, David I. Ríos-Vázquez, Arturo Liñan-Torres, Ramón Alberto Batista-García, Ernesto Pérez-Rueda, Norma Elena Rojas-Ruíz, and Sonia Dávila-Ramos. 2020. "The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities" Microorganisms 8, no. 11: 1677. https://doi.org/10.3390/microorganisms8111677
APA StyleCastelán-Sánchez, H. G., Meza-Rodríguez, P. M., Carrillo, E., Ríos-Vázquez, D. I., Liñan-Torres, A., Batista-García, R. A., Pérez-Rueda, E., Rojas-Ruíz, N. E., & Dávila-Ramos, S. (2020). The Microbial Composition in Circumneutral Thermal Springs from Chignahuapan, Puebla, Mexico Reveals the Presence of Particular Sulfur-Oxidizing Bacterial and Viral Communities. Microorganisms, 8(11), 1677. https://doi.org/10.3390/microorganisms8111677