Gut Microbiota and Immune System Interactions

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Gut Microbiota Metabolites (SCFAs)
2. Interactions between Gut Microbiota and Immune Cells
2.1. Immune Regulation of Gut Microbiota
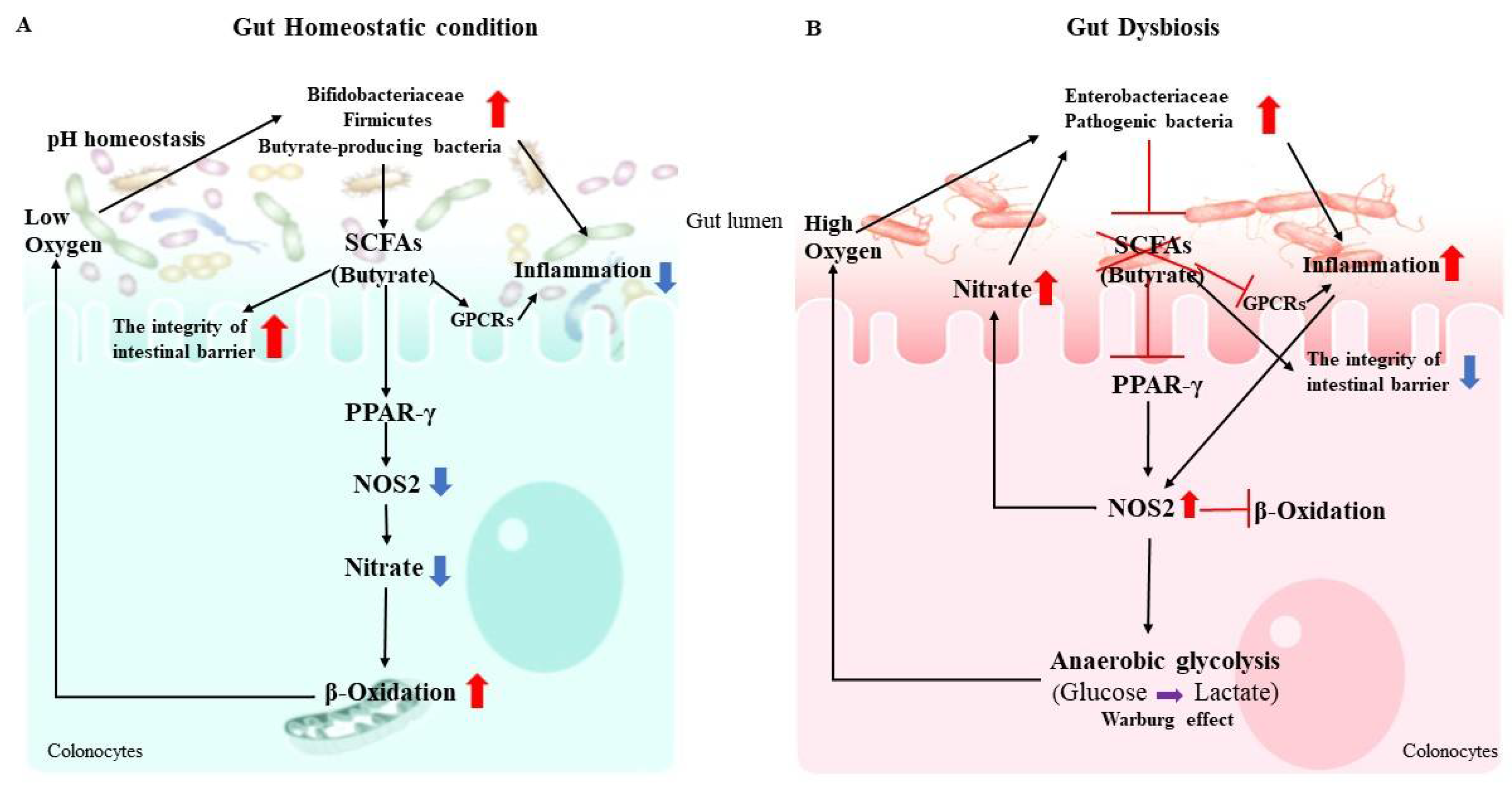
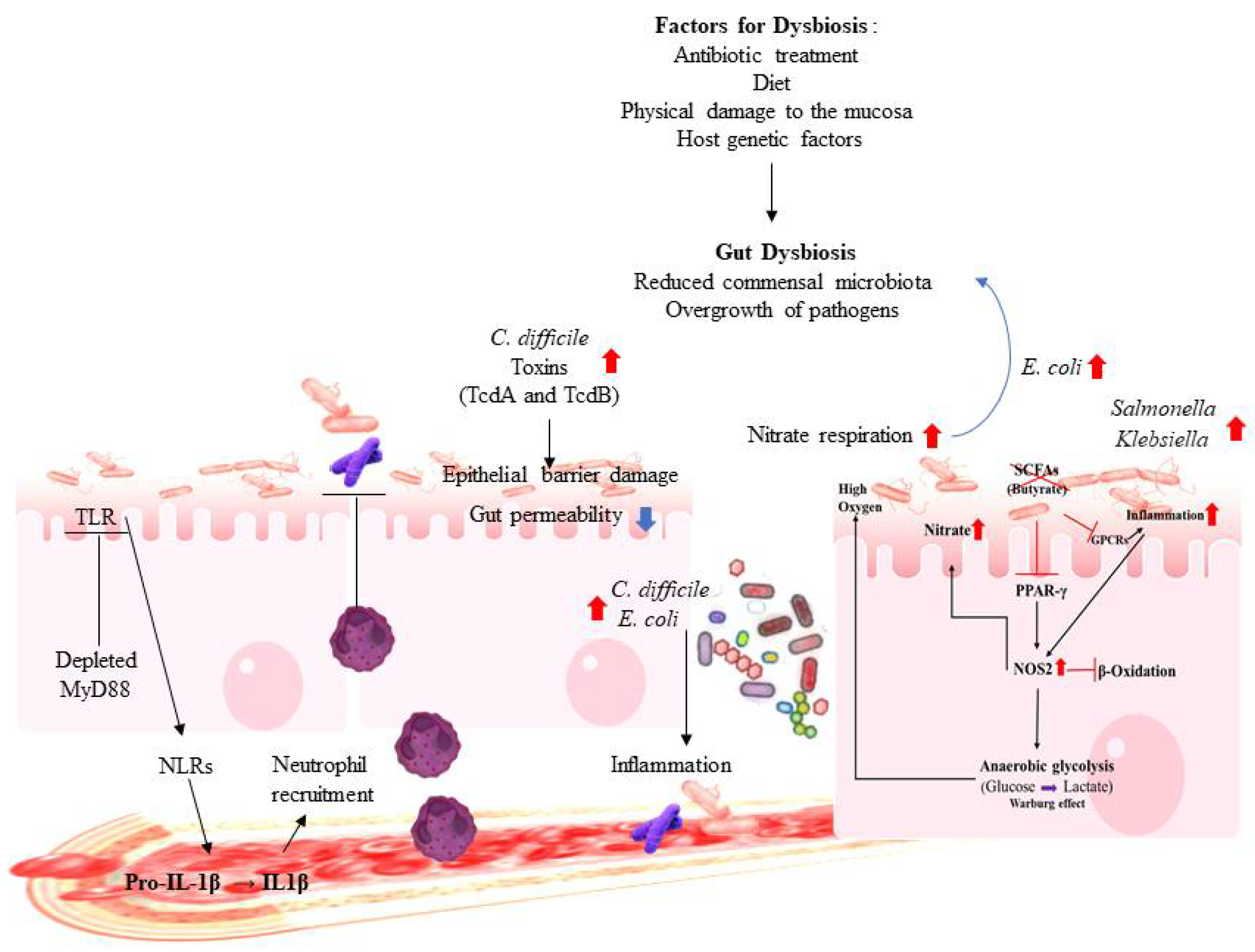
2.2. Gut Dysbiosis and Immune Dysregulation
3. Dysbiosis of the Gut Microbiota and Related Diseases
3.1. Gut Microbiota and Chronic Low-Grade Inflammation on Type 2 Diabetes (T2DM) and CVD
3.2. Gut Microbiota and Inflammatory Bowel Disease (IBD)
3.3. Gut Microbiota and Systemic Lupus Erythematosus (SLE)
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Venegas, D.P.; Marjorie, K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 43, 629–631. [Google Scholar] [CrossRef]
- Macfarlane, G.; Gibson, G. Microbiological aspects of the production of short-chain fatty acids in the large bowel. In Physiological and Clinical Aspects of Short-Chain Fatty Acids; Cambridge University Press: Cambridge, UK, 1995; pp. 87–105. [Google Scholar]
- Macfarlane, G.T.; Macfarlane, S. Human colonic microbiota: Ecology, physiology and metabolic potential of intestinal bacteria. Scand. J. Gastroenterol. Suppl. 1997, 222, 3–9. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Kaisar, M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate conditions human dendritic cells to prime type 1 regulatory T cells via both histone deacetylase inhibition and G protein-coupled receptor 109A signaling. Front. Immunol. 2017, 8, 1429. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A. GPR109A is a G-protein–coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Déjardin, F.; Sparwasser, T.; Bérard, M.; Cerf-Bensussan, N.; Eberl, G. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 2019, 50, 1276–1288.e1275. [Google Scholar] [CrossRef]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef]
- Lukasova, M.; Malaval, C.; Gille, A.; Kero, J.; Offermanns, S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J. Clin. Investig. 2011, 121, 1163–1173. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Hatanaka, E.; Sato, F.T.; Sampaio, S.C.; Curi, R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 2011, 22, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, H.; Chen, P.; Xie, H.; Tao, Y. Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal Transduct. Target. Ther. 2019, 4, 41. [Google Scholar] [CrossRef]
- Licciardi, P.V.; Ververis, K.; Karagiannis, T.C. Histone deacetylase inhibition and dietary short-chain Fatty acids. ISRN Allergy 2011, 2011, 869647. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Zhang, H.; Du, M.; Yang, Q.; Zhu, M.-J. Butyrate suppresses murine mast cell proliferation and cytokine production through inhibiting histone deacetylase. J. Nutr. Biochem. 2016, 27, 299–306. [Google Scholar] [CrossRef]
- Usami, M.; Kishimoto, K.; Ohata, A.; Miyoshi, M.; Aoyama, M.; Fueda, Y.; Kotani, J. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 2008, 28, 321–328. [Google Scholar] [CrossRef]
- Sellmer, A.; Stangl, H.; Beyer, M.; Grunstein, E.; Leonhardt, M.; Pongratz, H.; Eichhorn, E.; Elz, S.; Striegl, B.; Jenei-Lanzl, Z.; et al. Marbostat-100 Defines a New Class of Potent and Selective Antiinflammatory and Antirheumatic Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2018, 61, 3454–3477. [Google Scholar] [CrossRef]
- Mathew, O.P.; Ranganna, K.; Yatsu, F.M. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1-specific cell cycle proteins in vascular smooth muscle cells. Biomed. Pharmacother. 2010, 64, 733–740. [Google Scholar] [CrossRef]
- Itzhak, Y.; Liddie, S.; Anderson, K.L. Sodium butyrate-induced histone acetylation strengthens the expression of cocaine-associated contextual memory. Neurobiol. Learn. Mem. 2013, 102, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Vieira, E.L.; Leonel, A.J.; Sad, A.P.; Beltrao, N.R.; Costa, T.F.; Ferreira, T.M.; Gomes-Santos, A.C.; Faria, A.M.; Peluzio, M.C.; Cara, D.C.; et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J. Nutr. Biochem. 2012, 23, 430–436. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Béguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottière, H.M.; Lapaque, N. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Lamichane, S.; Dahal Lamichane, B.; Kwon, S.-M. Pivotal roles of peroxisome proliferator-activated receptors (PPARs) and their signal cascade for cellular and whole-body energy homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; Gerding, A.; van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the Metabolic Syndrome by Guar Gum-Derived Short-Chain Fatty Acids Depends on Peroxisome Proliferator-Activated Receptor gamma and Glucagon-Like Peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef]
- Athman, R.; Philpott, D. Innate immunity via Toll-like receptors and Nod proteins. Curr. Opin. Microbiol. 2004, 7, 25–32. [Google Scholar] [CrossRef]
- Oviedo-Boyso, J.; Bravo-Patiño, A.; Baizabal-Aguirre, V.M. Collaborative action of Toll-like and NOD-like receptors as modulators of the inflammatory response to pathogenic bacteria. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef]
- Sultani, M.; Stringer, A.M.; Bowen, J.M.; Gibson, R.J. Anti-inflammatory cytokines: Important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother. Res. Pract. 2012, 2012, 490804. [Google Scholar] [CrossRef]
- Iraporda, C.; Errea, A.; Romanin, D.E.; Cayet, D.; Pereyra, E.; Pignataro, O.; Sirard, J.C.; Garrote, G.L.; Abraham, A.G.; Rumbo, M. Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 2015, 220, 1161–1169. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, M.; Yang, X.; Hong, N.; Yu, C. Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J. Crohns. Colitis 2013, 7, e558–e568. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- El Asmar, R.; Panigrahi, P.; Bamford, P.; Berti, I.; Not, T.; Coppa, G.V.; Catassi, C.; Fasano, A. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology 2002, 123, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, D.H.; Erofeev, A.; Sandvik, A.; Grcic, V.; Jahnsen, F.L.; Gaustad, P.; McCoy, K.D.; Macpherson, A.J.; Meza-Zepeda, L.A.; Johansen, F.E. Depletion of murine intestinal microbiota: Effects on gut mucosa and epithelial gene expression. PLoS ONE 2011, 6, e17996. [Google Scholar] [CrossRef]
- Cullender, T.C.; Chassaing, B.; Janzon, A.; Kumar, K.; Muller, C.E.; Werner, J.J.; Angenent, L.T.; Bell, M.E.; Hay, A.G.; Peterson, D.A.; et al. Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 2013, 14, 571–581. [Google Scholar] [CrossRef]
- Atif, S.M.; Uematsu, S.; Akira, S.; McSorley, S.J. CD103-CD11b+ dendritic cells regulate the sensitivity of CD4 T-cell responses to bacterial flagellin. Mucosal. Immunol. 2014, 7, 68–77. [Google Scholar] [CrossRef]
- Neal, M.D.; Leaphart, C.; Levy, R.; Prince, J.; Billiar, T.R.; Watkins, S.; Li, J.; Cetin, S.; Ford, H.; Schreiber, A.; et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 2006, 176, 3070–3079. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Tsujibe, S.; Kiyoshima-Shibata, J.; Watanabe, Y.; Kato-Nagaoka, N.; Shida, K.; Matsumoto, S. Microbiota of the small intestine is selectively engulfed by phagocytes of the lamina propria and Peyer’s patches. PLoS ONE 2016, 11, e0163607. [Google Scholar] [CrossRef] [PubMed]
- Nerren, J.R.; He, H.; Genovese, K.; Kogut, M.H. Expression of the avian-specific toll-like receptor 15 in chicken heterophils is mediated by gram-negative and gram-positive bacteria, but not TLR agonists. Vet. Immunol. Immunopathol. 2010, 136, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Garate, I.; Garcia-Bueno, B.; Madrigal, J.L.; Bravo, L.; Berrocoso, E.; Caso, J.R.; Mico, J.A.; Leza, J.C. Origin and consequences of brain Toll-like receptor 4 pathway stimulation in an experimental model of depression. J. Neuroinflamm. 2011, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, M. Structural modifications of bacterial lipopolysaccharide that facilitate gram-negative bacteria evasion of host innate immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Cress, B.F.; Englaender, J.A.; He, W.; Kasper, D.; Linhardt, R.J.; Koffas, M.A. Masquerading microbial pathogens: Capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol. Rev. 2014, 38, 660–697. [Google Scholar] [CrossRef] [PubMed]
- Mostov, K.E. Transepithelial transport of immunoglobulins. Annu. Rev. Immunol. 1994, 12, 63–84. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef]
- Fernandez, M.I.; Sansonetti, P.J. Shigella interaction with intestinal epithelial cells determines the innate immune response in shigellosis. Int. J. Med. Microbiol. 2003, 293, 55–67. [Google Scholar] [CrossRef]
- Corthésy, B.; Benureau, Y.; Perrier, C.; Fourgeux, C.; Parez, N.; Greenberg, H.; Schwartz-Cornil, I. Rotavirus anti-VP6 secretory immunoglobulin A contributes to protection via intracellular neutralization but not via immune exclusion. J. Virol. 2006, 80, 10692–10699. [Google Scholar] [CrossRef]
- Mantis, N.J.; Cheung, M.C.; Chintalacharuvu, K.R.; Rey, J.; Corthesy, B.; Neutra, M.R. Selective adherence of IgA to murine Peyer’s patch M cells: Evidence for a novel IgA receptor. J. Immunol. 2002, 169, 1844–1851. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef]
- Kadaoui, K.A.; Corthesy, B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J. Immunol. 2007, 179, 7751–7757. [Google Scholar] [CrossRef]
- Boullier, S.; Tanguy, M.; Kadaoui, K.A.; Caubet, C.; Sansonetti, P.; Corthesy, B.; Phalipon, A. Secretory IgA-mediated neutralization of Shigella flexneri prevents intestinal tissue destruction by down-regulating inflammatory circuits. J. Immunol. 2009, 183, 5879–5885. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Honda, K. Intestinal commensal microbes as immune modulators. Cell Host Microbe 2012, 12, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.P.; Hawrelak, J. The causes of intestinal dysbiosis: A review. Altern. Med. Rev. 2004, 9, 180–197. [Google Scholar]
- Wang, B.; Yao, M.; Lv, L.; Ling, Z.; Li, L. The human microbiota in health and disease. Engineering 2017, 3, 71–82. [Google Scholar] [CrossRef]
- Gillis, C.C.; Hughes, E.R.; Spiga, L.; Winter, M.G.; Zhu, W.; de Carvalho, T.F.; Chanin, R.B.; Behrendt, C.L.; Hooper, L.V.; Santos, R.L. Dysbiosis-associated change in host metabolism generates lactate to support Salmonella growth. Cell Host Microbe 2018, 23, 54–64.e56. [Google Scholar] [CrossRef]
- Kosiewicz, M.M.; Zirnheld, A.L.; Alard, P. Gut microbiota, immunity, and disease: A complex relationship. Front. Microbiol. 2011, 2, 180. [Google Scholar] [CrossRef]
- Cervantes, J.L.; Hong, B.Y. Dysbiosis and Immune Dysregulation in Outer Space. Int. Rev. Immunol. 2016, 35, 67–82. [Google Scholar] [CrossRef]
- Douglas, C.A.; Ivey, K.L.; Papanicolas, L.E.; Best, K.P.; Muhlhausler, B.S.; Rogers, G.B. DNA extraction approaches substantially influence the assessment of the human breast milk microbiome. Sci. Rep. 2020, 10, 123. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.R.; Sartor, R.B. The role of diet on intestinal microbiota metabolism: Downstream impacts on host immune function and health, and therapeutic implications. J. Gastroenterol. 2014, 49, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Liu, X.; Cheng, Y.; Jiang, X.; Jiang, H.; Wang, Y.; Li, L. Decreased Diversity of the Oral Microbiota of Patients with Hepatitis B Virus-Induced Chronic Liver Disease: A Pilot Project. Sci. Rep. 2015, 5, 17098. [Google Scholar] [CrossRef]
- Gu, S.; Chen, Y.; Zhang, X.; Lu, H.; Lv, T.; Shen, P.; Lv, L.; Zheng, B.; Jiang, X.; Li, L. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. 2016, 18, 30–38. [Google Scholar] [CrossRef]
- Everard, A.; Cani, P.D. Diabetes, obesity and gut microbiota. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef]
- Miele, L.; Giorgio, V.; Alberelli, M.A.; De Candia, E.; Gasbarrini, A.; Grieco, A. Impact of Gut Microbiota on Obesity, Diabetes, and Cardiovascular Disease Risk. Curr. Cardiol. Rep. 2015, 17, 120. [Google Scholar] [CrossRef]
- Sanz, Y.; Santacruz, A.; Gauffin, P. Gut microbiota in obesity and metabolic disorders. Proc. Nutr. Soc. 2010, 69, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef]
- Garvey, W.T.; Maianu, L.; Zhu, J.-H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef]
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Krssak, M.; Roden, M. The role of lipid accumulation in liver and muscle for insulin resistance and type 2 diabetes mellitus in humans. Rev. Endocr. Metab. Disord. 2004, 5, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Snel, M.; Jonker, J.T.; Schoones, J.; Lamb, H.; de Roos, A.; Pijl, H.; Smit, J.W.; Meinders, A.E.; Jazet, I.M. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol. 2012, 2012, 983814. [Google Scholar] [CrossRef]
- Kurokawa, J.; Arai, S.; Nakashima, K.; Nagano, H.; Nishijima, A.; Miyata, K.; Ose, R.; Mori, M.; Kubota, N.; Kadowaki, T. Macrophage-derived AIM is endocytosed into adipocytes and decreases lipid droplets via inhibition of fatty acid synthase activity. Cell Metab. 2010, 11, 479–492. [Google Scholar] [CrossRef]
- Lampidonis, A.D.; Rogdakis, E.; Voutsinas, G.E.; Stravopodis, D.J. The resurgence of Hormone-Sensitive Lipase (HSL) in mammalian lipolysis. Gene 2011, 477, 1–11. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, S.S. Probiotics and Prebiotics: Present Status and Future Perspectives on Metabolic Disorders. Nutrients 2016, 8, 173. [Google Scholar] [CrossRef]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M.; et al. Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high fat diet-fed mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sorensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Meijer, K.; de Vos, P.; Priebe, M.G. Butyrate and other short-chain fatty acids as modulators of immunity: What relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.; Lee, J.H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef]
- Nagao-Kitamoto, H.; Shreiner, A.B.; Gillilland, M.G., 3rd; Kitamoto, S.; Ishii, C.; Hirayama, A.; Kuffa, P.; El-Zaatari, M.; Grasberger, H.; Seekatz, A.M.; et al. Functional Characterization of Inflammatory Bowel Disease-Associated Gut Dysbiosis in Gnotobiotic Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 468–481. [Google Scholar] [CrossRef]
- Scher, J.U.; Ubeda, C.; Artacho, A.; Attur, M.; Isaac, S.; Reddy, S.M.; Marmon, S.; Neimann, A.; Brusca, S.; Patel, T.; et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015, 67, 128–139. [Google Scholar] [CrossRef]
- Kaur, N.; Chen, C.C.; Luther, J.; Kao, J.Y. Intestinal dysbiosis in inflammatory bowel disease. Gut Microbes 2011, 2, 211–216. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Goboes, K.; Peeters, M.; Hiele, M.; Penninckx, F.; Aerts, R.; Kerremans, R.; Vantrappen, G. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991, 338, 771–774. [Google Scholar] [CrossRef]
- D’Haens, G.R.; Geboes, K.; Peeters, M.; Baert, F.; Penninckx, F.; Rutgeerts, P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 1998, 114, 262–267. [Google Scholar] [CrossRef]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.R.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.E.; Thiennimitr, P.; Winter, M.G.; Butler, B.P.; Huseby, D.L.; Crawford, R.W.; Russell, J.M.; Bevins, C.L.; Adams, L.G.; Tsolis, R.M.; et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 2010, 467, 426–429. [Google Scholar] [CrossRef]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, e244. [Google Scholar] [CrossRef]
- O’Hara, J.R.; Feener, T.D.; Fischer, C.D.; Buret, A.G. Campylobacter jejuni disrupts protective Toll-like receptor 9 signaling in colonic epithelial cells and increases the severity of dextran sulfate sodium-induced colitis in mice. Infect. Immun. 2012, 80, 1563–1571. [Google Scholar] [CrossRef]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Gladman, D.D.; Ibanez, D.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- He, Z.; Shao, T.; Li, H.; Xie, Z.; Wen, C. Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut Pathog. 2016, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.M.; Edwards, M.R.; Mu, Q.; Yu, Y.; Vieson, M.D.; Reilly, C.M.; Ahmed, S.A.; Bankole, A.A. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.; de Paz, B.; Rodriguez-Carrio, J.; Hevia, A.; Sanchez, B.; Margolles, A.; Suarez, A. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci. Rep. 2016, 6, 24072. [Google Scholar] [CrossRef]
- Wolf, S.J.; Estadt, S.N.; Theros, J.; Moore, T.; Ellis, J.; Liu, J.; Reed, T.J.; Jacob, C.O.; Gudjonsson, J.E.; Kahlenberg, J.M. Ultraviolet light induces increased T cell activation in lupus-prone mice via type I IFN-dependent inhibition of T regulatory cells. J. Autoimmun. 2019, 103, 102291. [Google Scholar] [CrossRef]
- Sontheimer, C.; Liggitt, D.; Elkon, K.B. Ultraviolet B Irradiation Causes Stimulator of Interferon Genes-Dependent Production of Protective Type I Interferon in Mouse Skin by Recruited Inflammatory Monocytes. Arthritis Rheumatol. 2017, 69, 826–836. [Google Scholar] [CrossRef]
- Båve, U.; Magnusson, M.; Eloranta, M.-L.; Perers, A.; Alm, G.V.; Rönnblom, L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J. Immunol. 2003, 171, 3296–3302. [Google Scholar] [CrossRef]
- Bolland, S.; Ravetch, J.V. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity 2000, 13, 277–285. [Google Scholar] [CrossRef]
- Li, X.; Ptacek, T.S.; Brown, E.E.; Edberg, J.C. Fcγ receptors: Structure, function and role as genetic risk factors in SLE. Genes Immun. 2009, 10, 380–389. [Google Scholar] [CrossRef]
- Scalapino, K.J.; Tang, Q.; Bluestone, J.A.; Bonyhadi, M.L.; Daikh, D.I. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J. Immunol. 2006, 177, 1451–1459. [Google Scholar] [CrossRef]
- Denny, M.F.; Yalavarthi, S.; Zhao, W.; Thacker, S.G.; Anderson, M.; Sandy, A.R.; McCune, W.J.; Kaplan, M.J. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J. Immunol. 2010, 184, 3284–3297. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Rosser, E.C.; Oleinika, K.; Tonon, S.; Doyle, R.; Bosma, A.; Carter, N.A.; Harris, K.A.; Jones, S.A.; Klein, N.; Mauri, C. Regulatory B cells are induced by gut microbiota-driven interleukin-1beta and interleukin-6 production. Nat. Med. 2014, 20, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Tavella, V.J.; Kirby, J.L.; Cecere, T.E.; Chung, M.; Lee, J.; Li, S.; Ahmed, S.A.; Eden, K.; Allen, I.C.; et al. Antibiotics ameliorate lupus-like symptoms in mice. Sci. Rep. 2017, 7, 13675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liao, X.; Sparks, J.B.; Luo, X.M. Dynamics of gut microbiota in autoimmune lupus. Appl. Environ. Microbiol. 2014, 80, 7551–7560. [Google Scholar] [CrossRef] [PubMed]
- Mu, Q.; Zhang, H.; Liao, X.; Lin, K.; Liu, H.; Edwards, M.R.; Ahmed, S.A.; Yuan, R.; Li, L.; Cecere, T.E.; et al. Control of lupus nephritis by changes of gut microbiota. Microbiome 2017, 5, 73. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.S.; Eisenberg, R.A.; Theofilopoulos, A.; Izui, S.; Wilson, C.B.; McConahey, P.; Murphy, E.D.; Roths, J.B.; Dixon, F. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978, 148, 1198–1215. [Google Scholar] [CrossRef]
- Rodríguez-Carrio, J.; López, P.; Sánchez, B.; González, S.; Gueimonde, M.; Margolles, A.; de Los Reyes-Gavilán, C.G.; Suárez, A. Intestinal dysbiosis is associated with altered short-chain fatty acids and serum-free fatty acids in systemic lupus erythematosus. Front. Immunol. 2017, 8, 23. [Google Scholar] [CrossRef]
- Zhang, Q.; Yin, X.; Wang, H.; Wu, X.; Li, X.; Li, Y.; Zhang, X.; Fu, C.; Li, H.; Qiu, Y. Fecal Metabolomics and Potential Biomarkers for Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 976. [Google Scholar] [CrossRef]
- Zhang, T.; Mohan, C. Caution in studying and interpreting the lupus metabolome. Arthritis Res. Ther. 2020, 22, 172. [Google Scholar] [CrossRef] [PubMed]
- Eichhorst, A.; Schäfer, A.-L.; Voll, R.E.; Chevalier, N. P55 Influence of dietary fibre and short-chain fatty acids on the pathogenesis of systemic lupus erythematosus. Lupus Sci. Med. 2020, 7, 55. [Google Scholar] [CrossRef]
- Gu, J.; Mao, B.; Cui, S.; Liu, X.; Zhang, H.; Zhao, J.; Chen, W. Metagenomic insights into the effects of fructooligosaccharides (FOS) on the composition of luminal and mucosal microbiota in C57BL/6J mice, especially the Bifidobacterium composition. Nutrients 2019, 11, 2431. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; You, H.J.; Lee, Y.-G.; Jeong, Y.; Johnston, T.V.; Baek, N.-I.; Ku, S.; Ji, G.E. Production, Structural Characterization, and In Vitro Assessment of the Prebiotic Potential of Butyl-Fructooligosaccharides. Int. J. Mol. Sci. 2020, 21, 445. [Google Scholar] [CrossRef]
- Kusumo, P.D.; Maulahela, H.; Utari, A.P.; Surono, I.S.; Soebandrio, A.; Abdullah, M. Probiotic Lactobacillus plantarum IS 10506 supplementation increase SCFA of women with functional constipation. Iran. J. Microbiol. 2019, 11, 389. [Google Scholar] [PubMed]
- Bhutia, Y.D.; Ganapathy, V. Short, but smart: SCFAs train T cells in the gut to fight autoimmunity in the brain. Immunity 2015, 43, 629–631. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. https://doi.org/10.3390/microorganisms8101587
Yoo JY, Groer M, Dutra SVO, Sarkar A, McSkimming DI. Gut Microbiota and Immune System Interactions. Microorganisms. 2020; 8(10):1587. https://doi.org/10.3390/microorganisms8101587
Chicago/Turabian StyleYoo, Ji Youn, Maureen Groer, Samia Valeria Ozorio Dutra, Anujit Sarkar, and Daniel Ian McSkimming. 2020. "Gut Microbiota and Immune System Interactions" Microorganisms 8, no. 10: 1587. https://doi.org/10.3390/microorganisms8101587
APA StyleYoo, J. Y., Groer, M., Dutra, S. V. O., Sarkar, A., & McSkimming, D. I. (2020). Gut Microbiota and Immune System Interactions. Microorganisms, 8(10), 1587. https://doi.org/10.3390/microorganisms8101587