Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
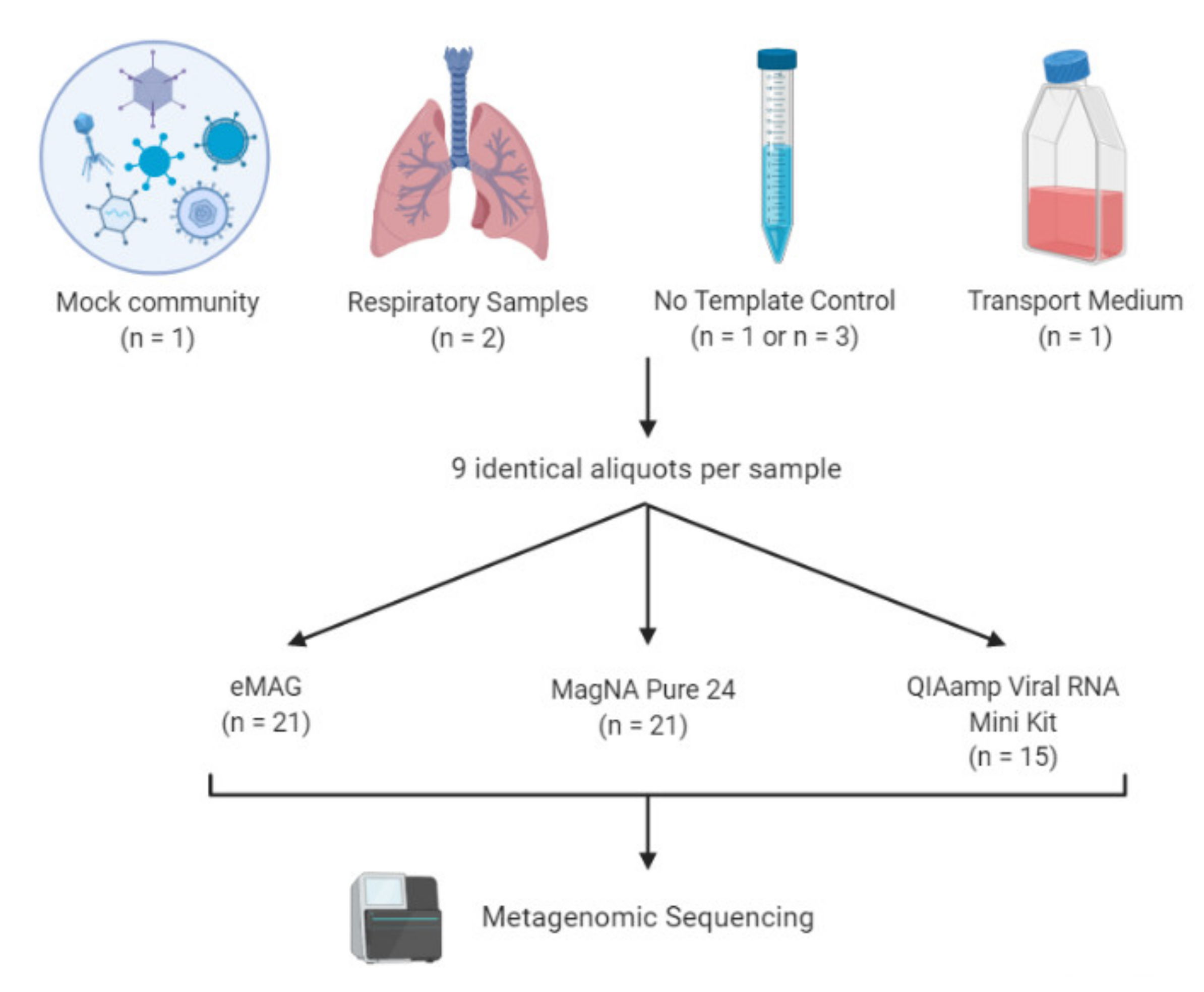
2.1. Design of the Mock Virome
2.2. Sample Collection
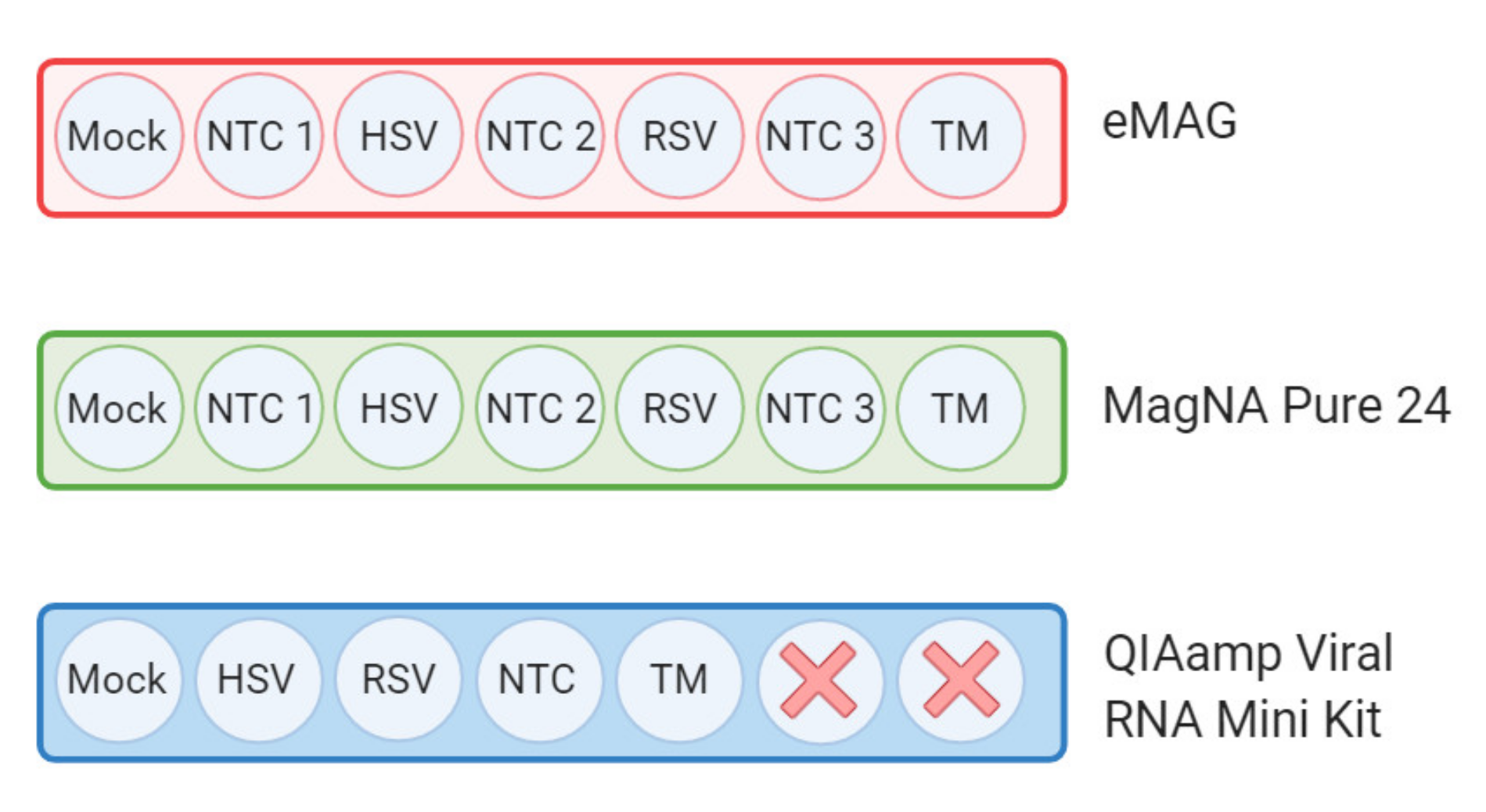
2.3. Nucleic Acid Extraction
2.4. Metagenomic Workflow
2.5. Bioinformatic Analysis
2.6. Statistical Analyses
2.7. Data Availability
2.8. Ethics
3. Results
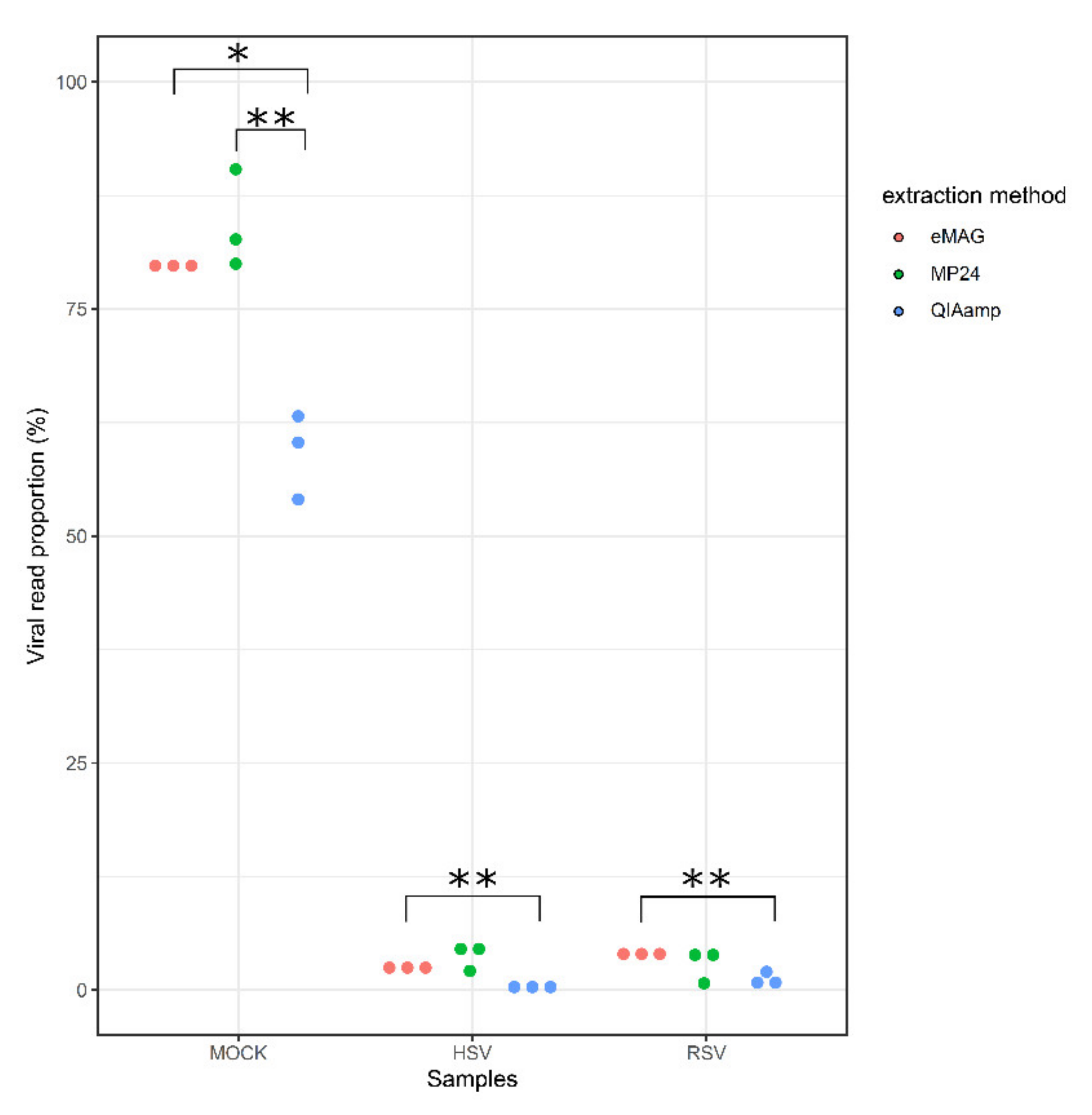
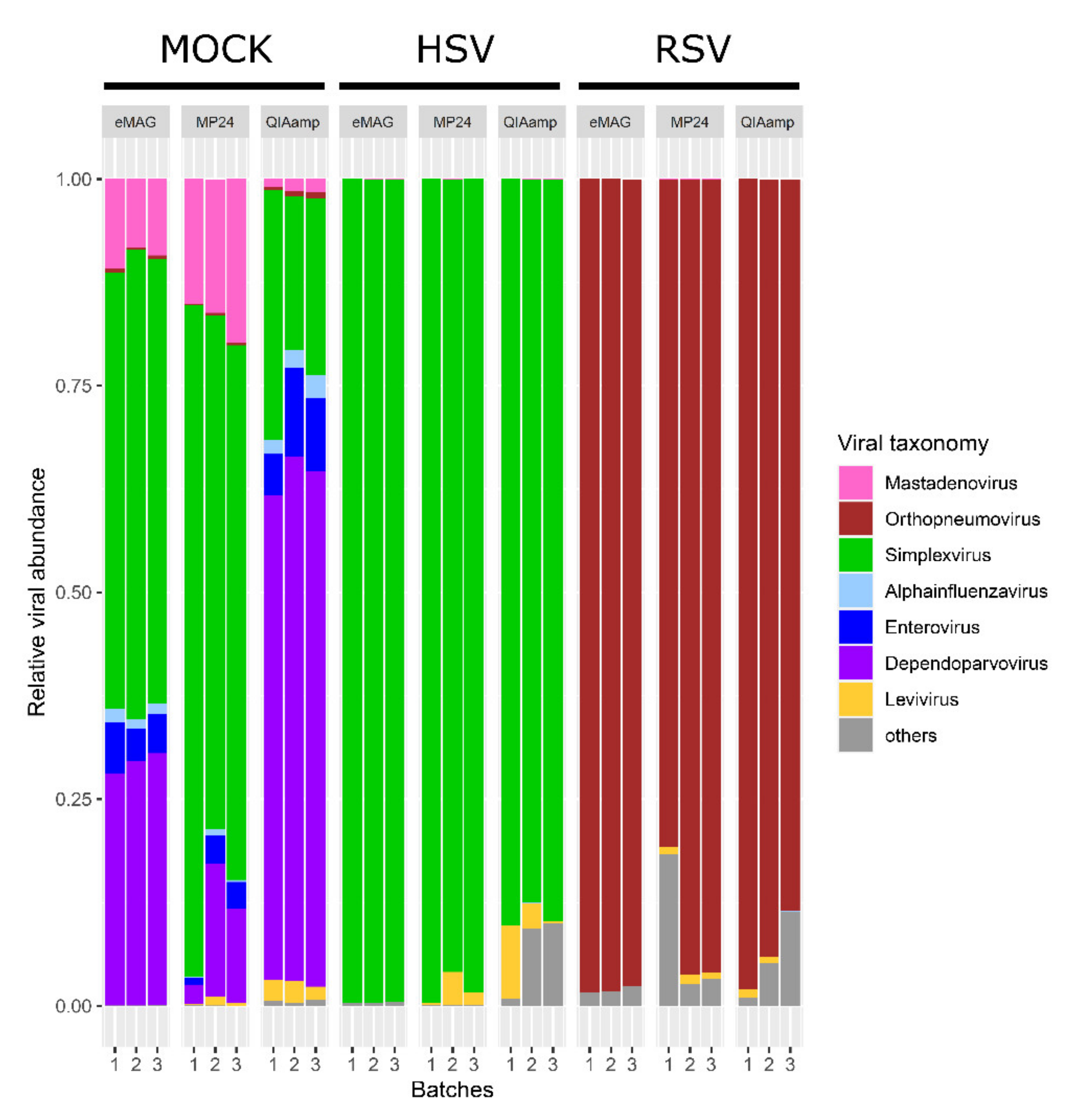
3.1. Sensitivity for the Detection of the Targeted Viruses
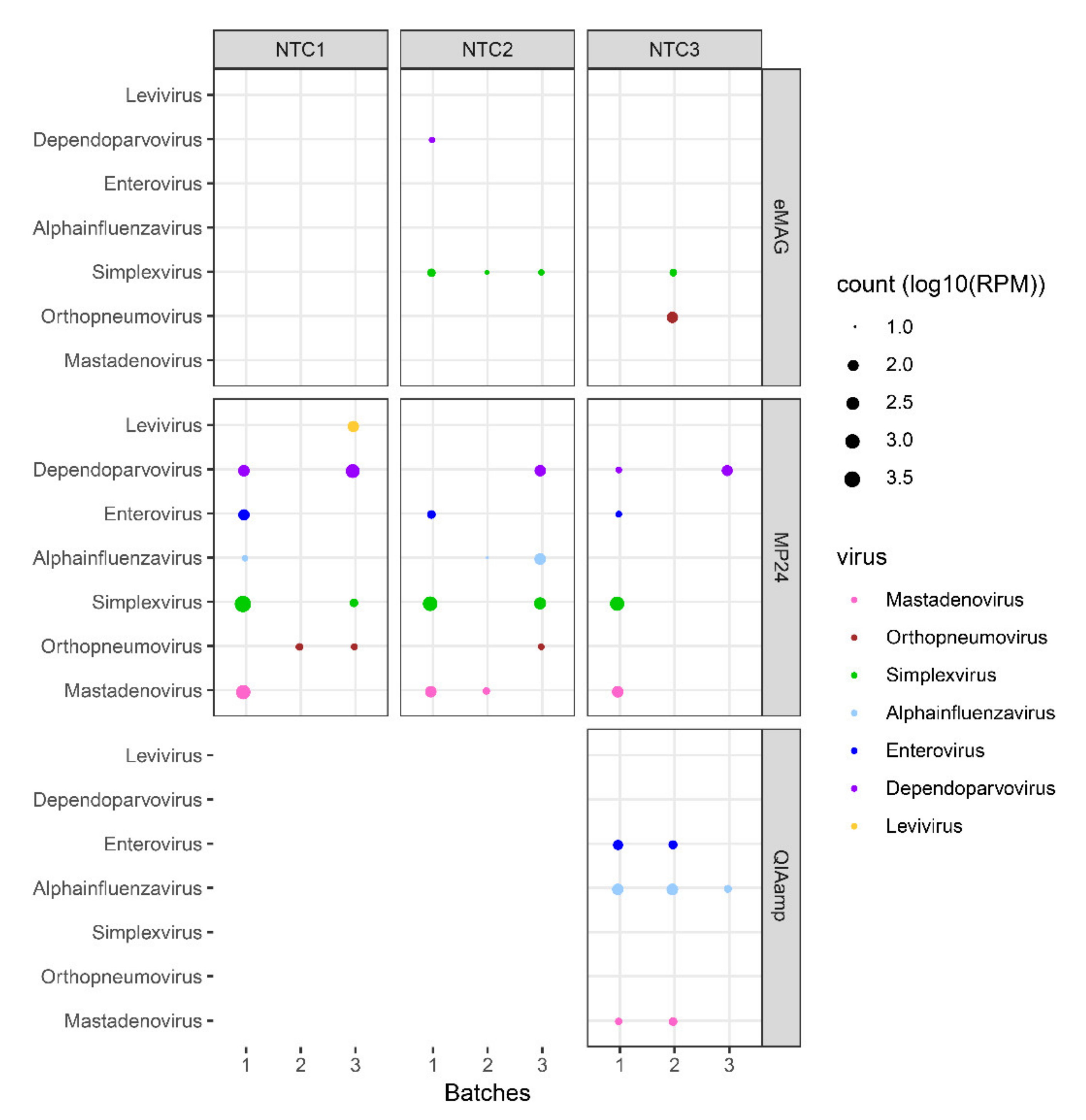
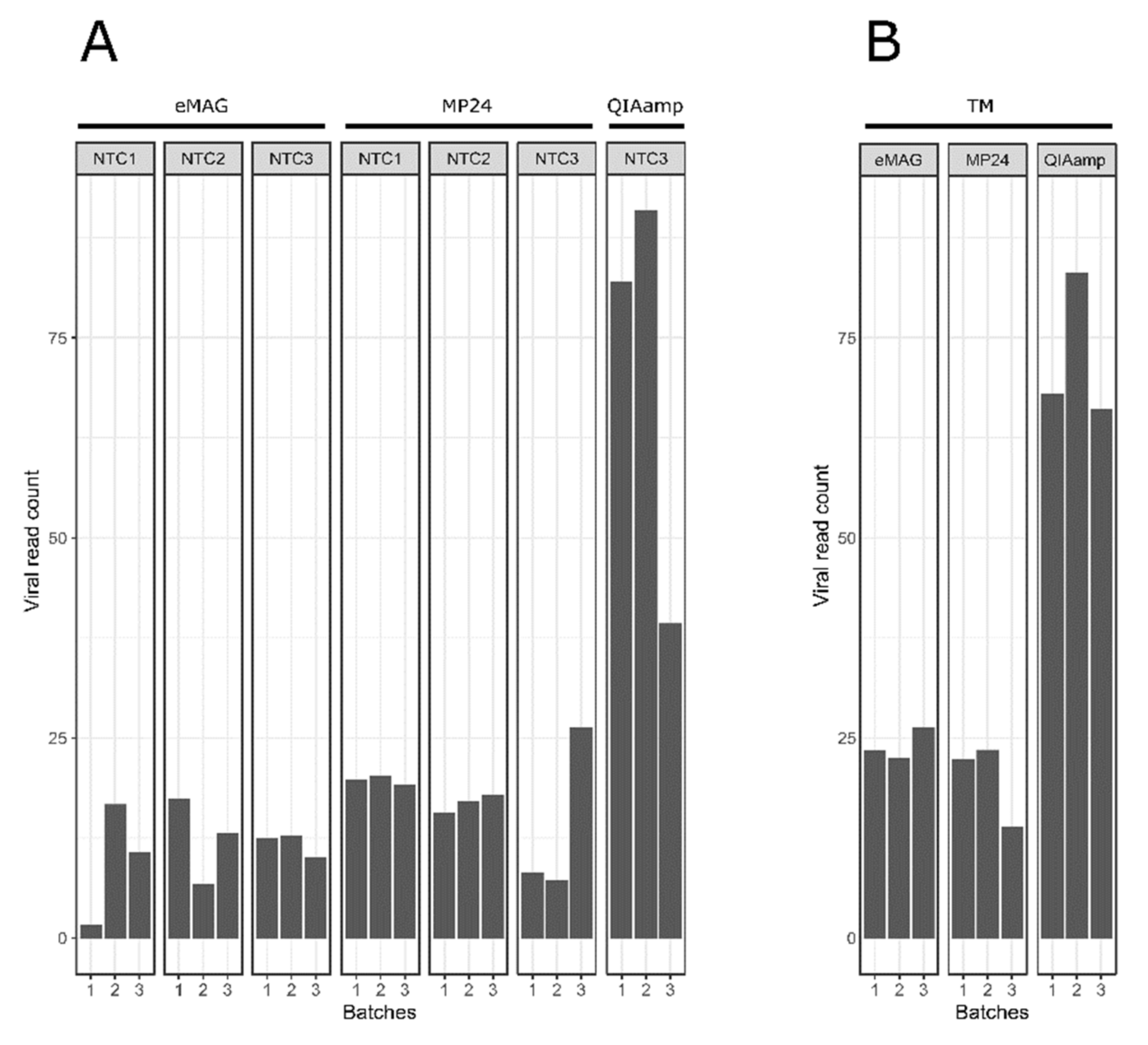
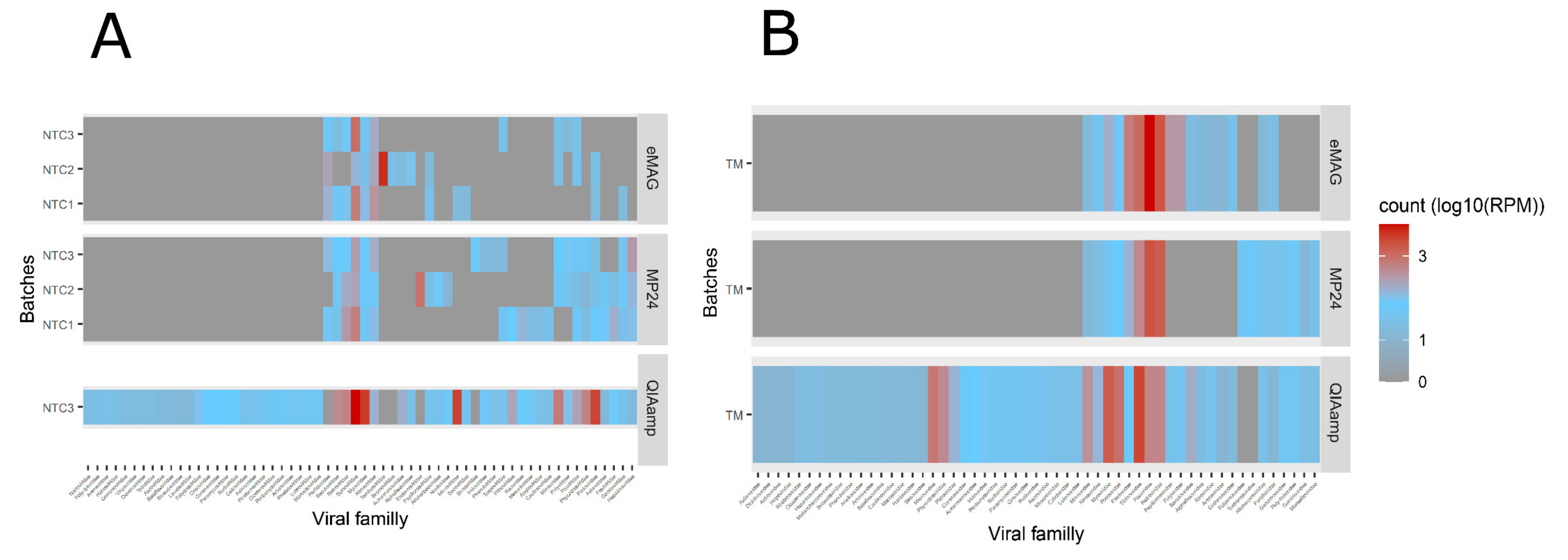
3.2. Sample Cross-Contamination
3.3. Kitome Assessment
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef]
- Xu, L.; Zhu, Y.; Ren, L.; Xu, B.; Liu, C.; Xie, Z.; Shen, K. Characterization of the nasopharyngeal viral microbiome from children with community-acquired pneumonia but negative for Luminex xTAG respiratory viral panel assay detection. J. Med. Virol. 2017, 89, 2098–2107. [Google Scholar] [CrossRef]
- Yang, Y.; Walls, S.D.; Gross, S.M.; Schroth, G.P.; Jarman, R.G.; Hang, J. Targeted Sequencing of Respiratory Viruses in Clinical Specimens for Pathogen Identification and Genome-Wide Analysis. In The Human Virome; Moya, A., Pérez Brocal, V., Eds.; Springer: New York, NY, USA, 2018; Volume 1838, pp. 125–140. [Google Scholar] [CrossRef]
- Cummings, M.J.; Tokarz, R.; Bakamutumaho, B.; Kayiwa, J.; Byaruhanga, T.; Owor, N.; Namagambo, B.; Wolf, A.; Mathema, B.; Lutwama, J.J.; et al. Precision Surveillance for Viral Respiratory Pathogens: Virome Capture Sequencing for the Detection and Genomic Characterization of Severe Acute Respiratory Infection in Uganda. Clin. Infect. Dis. 2019, 68, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Paskey, A.C.; Frey, K.G.; Schroth, G.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment post-library preparation enhances the sensitivity of high-throughput sequencing-based detection and characterization of viruses from complex samples. BMC Genom. 2019, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Kufner, V.; Plate, A.; Schmutz, S.; Braun, D.L.; Günthard, H.F.; Capaul, R.; Zbinden, A.; Mueller, N.J.; Trkola, A.; Huber, M. Two Years of Viral Metagenomics in a Tertiary Diagnostics Unit: Evaluation of the First 105 Cases. Genes 2019, 10, 661. [Google Scholar] [CrossRef]
- Eibach, D.; Hogan, B.; Sarpong, N.; Winter, D.; Struck, N.S.; Adu-Sarkodie, Y.; Owusu-Dabo, E.; Schmidt-Chanasit, J.; May, J.; Cadar, D. Viral metagenomics revealed novel betatorquevirus species in pediatric inpatients with encephalitis/meningoencephalitis from Ghana. Sci. Rep. 2019, 9, 2360. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, W.; Zhang, Q.; Xu, K.; Ye, G.; Wu, W.; Sun, Z.; Liu, F.; Wu, K.; Zhong, B.; et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 2020, 9, 313–319. [Google Scholar] [CrossRef]
- Moore, N.E.; Wang, J.; Hewitt, J.; Croucher, D.; Williamson, D.A.; Paine, S.; Yen, S.; Greening, G.E.; Hall, R.J. Metagenomic Analysis of Viruses in Feces from Unsolved Outbreaks of Gastroenteritis in Humans. J. Clin. Microbiol. 2015, 53, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Silins, R.; Castro-Mejía, J.L.; Kot, W.; Jessen, L.; Thorsen, J.; Shah, S.; Stokholm, J.; Bisgaard, H.; Moineau, S.; et al. A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses 2019, 11, 667. [Google Scholar] [CrossRef]
- Law, J.; Jovel, J.; Patterson, J.; Ford, G.; O’keefe, S.; Wang, W.; Meng, B.; Song, D.; Zhang, Y.; Tian, Z.; et al. Identification of Hepatotropic Viruses from Plasma Using Deep Sequencing: A Next Generation Diagnostic Tool. PLoS ONE 2013, 8, e60595. [Google Scholar] [CrossRef]
- Rascovan, N.; Duraisamy, R.; Desnues, C. Metagenomics and the Human Virome in Asymptomatic Individuals. Annu. Rev. Microbiol. 2016, 70, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Naccache, S.N.; Samayoa, E.; Messacar, K.; Arevalo, S.; Federman, S.; Stryke, D.; Pham, E.; Fung, B.; Bolosky, W.J.; et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res 2019, 29, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C.; et al. Clinical Metagenomic Sequencing for Diagnosis of Meningitis and Encephalitis. New Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Bzhalava, D.; Ekström, J.; Hultin, E.; Dillner, J.; Forslund, O. Metagenomic sequencing of “HPV-negative” condylomas detects novel putative HPV types. Virology 2013, 440, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A. Protocol for Metagenomic Virus Detection in Clinical Specimens. Emerg. Infect. Dis. 2015, 21, 48–57. [Google Scholar] [CrossRef]
- Lysholm, F.; Wetterbom, A.; Lindau, C.; Darban, H.; Bjerkner, A.; Fahlander, K.; Lindberg, A.M.; Persson, B.; Allander, T.; Andersson, B. Characterization of the Viral Microbiome in Patients with Severe Lower Respiratory Tract Infections, Using Metagenomic Sequencing. PLoS ONE 2012, 7, e30875. [Google Scholar] [CrossRef]
- Fischer, N.; Indenbirken, D.; Meyer, T.; Lütgehetmann, M.; Lellek, H.; Spohn, M.; Aepfelbacher, M.; Alawi, M.; Grundhoff, A. Evaluation of Unbiased Next-Generation Sequencing of RNA (RNA-seq) as a Diagnostic Method in Influenza Virus-Positive Respiratory Samples. J. Clin. Microbiol. 2015, 53, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Kawada, J.; Horiba, K.; Okuno, Y.; Okumura, T.; Suzuki, T.; Torii, Y.; Kawabe, S.; Wada, S.; Ikeyama, T.; et al. Metagenomic analysis using next-generation sequencing of pathogens in bronchoalveolar lavage fluid from pediatric patients with respiratory failure. Sci. Rep. 2019, 9, 1209. [Google Scholar] [CrossRef]
- Li, Y.; Fu, X.; Ma, J.; Zhang, J.; Hu, Y.; Dong, W.; Wan, Z.; Li, Q.; Kuang, Y.-Q.; Lan, K.; et al. Altered respiratory virome and serum cytokine profile associated with recurrent respiratory tract infections in children. Nat. Commun. 2019, 10, 2288. [Google Scholar] [CrossRef]
- Van den Munckhof, E.H.A.; de Koning, M.N.C.; Quint, W.G.V.; van Doorn, L.-J.; Leverstein-van Hall, M.A. Evaluation of a stepwise approach using microbiota analysis, species-specific qPCRs and culture for the diagnosis of lower respiratory tract infections. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 747–754. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the Promises and Hurdles of Metagenomic Next-Generation Sequencing as a Diagnostic Tool for Infectious Diseases. Clin. Infect. Dis. 2018, 66, 778–788. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Zagordi, O.; Geissberger, F.-D.; Kufner, V.; Schmutz, S.; Böni, J.; Metzner, K.J.; Trkola, A.; Huber, M. Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 2017, 5, 94. [Google Scholar] [CrossRef] [PubMed]
- Knepp, J.H.; Geahr, M.A.; Forman, M.S.; Valsamakis, A. Comparison of Automated and Manual Nucleic Acid Extraction Methods for Detection of Enterovirus RNA. J. Clin. Microbiol. 2003, 41, 3532–3536. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Seet, H.; Khan, Y.; Wright, C.; Nadarajah, R. Comparison of QIAGEN Automated Nucleic Acid Extraction Methods for CMV Quantitative PCR Testing. Am. J. Clin. Pathol. 2010, 133, 558–563. [Google Scholar] [CrossRef]
- Verheyen, J.; Kaiser, R.; Bozic, M.; Timmen-Wego, M.; Maier, B.K.; Kessler, H.H. Extraction of viral nucleic acids: Comparison of five automated nucleic acid extraction platforms. J. Clin. Virol. 2012, 54, 255–259. [Google Scholar] [CrossRef]
- Lewandowski, K.; Bell, A.; Miles, R.; Carne, S.; Wooldridge, D.; Manso, C.; Hennessy, N.; Bailey, D.; Pullan, S.T.; Gharbia, S.; et al. The Effect of Nucleic Acid Extraction Platforms and Sample Storage on the Integrity of Viral RNA for Use in Whole Genome Sequencing. J. Mol. Diagn. 2017, 19, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Klenner, J.; Kohl, C.; Dabrowski, P.W.; Nitsche, A. Comparing Viral Metagenomic Extraction Methods. Curr. Issues Mol. Biol. 2017, 59–70. [Google Scholar] [CrossRef]
- Zhang, D.; Lou, X.; Yan, H.; Pan, J.; Mao, H.; Tang, H.; Shu, Y.; Zhao, Y.; Liu, L.; Li, J.; et al. Metagenomic analysis of viral nucleic acid extraction methods in respiratory clinical samples. BMC Genom. 2018, 19, 773. [Google Scholar] [CrossRef]
- Yang, J.; Yang, F.; Ren, L.; Xiong, Z.; Wu, Z.; Dong, J.; Sun, L.; Zhang, T.; Hu, Y.; Duet, J.; et al. Unbiased Parallel Detection of Viral Pathogens in Clinical Samples by Use of a Metagenomic Approach. J. Clin. Microbiol. 2011, 49, 3463–3469. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Payen, M.; Woerther, P.-L.; Rodriguez, C.; Botterel, F. Combined bacterial and fungal targeted amplicon sequencing of respiratory samples: Does the DNA extraction method matter? PLoS ONE 2020, 15, e0232215. [Google Scholar] [CrossRef]
- Sui, H.; Weil, A.A.; Nuwagira, E.; Qadri, F.; Ryan, E.T.; Mezzari, M.P.; Phipatanakul, W.; Lai, P.S. Impact of DNA Extraction Method on Variation in Human and Built Environment Microbial Community and Functional Profiles Assessed by Shotgun Metagenomics Sequencing. Front. Microbiol. 2020, 11, 953. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Jeraldo, P.; Greenwood-Quaintance, K.E.; Yao, J.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis. J. Clin. Microbiol. 2017, 55, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Drengenes, C.; Wiker, H.G.; Kalananthan, T.; Nordeide, E.; Eagan, T.M.L.; Nielsen, R. Laboratory contamination in airway microbiome studies. BMC Microbiol. 2019, 19, 187. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett Jr, J.; Delwart, E.L.; Chiu, C.Y. The Perils of Pathogen Discovery: Origin of a Novel Parvovirus-Like Hybrid Genome Traced to Nucleic Acid Extraction Spin Columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef]
- Kjartansdóttir, K.R.; Friis-Nielsen, J.; Asplund, M.; Mollerup, S.; Mourier, T.; Jensen, R.H.; Hansen, T.A.; Rey-Iglesia, A.; Richter, S.R.; Alquezar-Planas, D.E.; et al. Traces of ATCV-1 associated with laboratory component contamination. Proc. Natl. Acad. Sci. USA 2015, 112, E925–E926. [Google Scholar] [CrossRef]
- Stinson, L.F.; Keelan, J.A.; Payne, M.S. Identification and removal of contaminating microbial DNA from PCR reagents: Impact on low-biomass microbiome analyses. Lett. Appl. Microbiol. 2019, 68, 2–8. [Google Scholar] [CrossRef]
- Miller, R.R.; Uyaguari-Diaz, M.; McCabe, M.N.; Montoya, V.; Gardy, J.L.; Parker, S.; Steiner, T.; Hsiao, W.; Nesbitt, M.J.; Tang, P.; et al. Metagenomic Investigation of Plasma in Individuals with ME/CFS Highlights the Importance of Technical Controls to Elucidate Contamination and Batch Effects. PLoS ONE 2016, 11, e0165691. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assuring the Quality of Next-Generation Sequencing in Clinical Microbiology and Public Health Laboratories. J. Clin. Microbiol. 2016, 54, 2857–2865. [Google Scholar] [CrossRef]
- Schlaberg, R.; Queen, K.; Simmon, K.; Tardif, K.; Stockmann, C.; Flygare, S.; Kennedy, B.; Voelkerding, K.; Bramley, A.; Zhang, J.; et al. Viral Pathogen Detection by Metagenomics and Pan-Viral Group Polymerase Chain Reaction in Children With Pneumonia Lacking Identifiable Etiology. J. Infect. Dis. 2017, 215, 1407–1415. [Google Scholar] [CrossRef]
- Li, L.; Deng, X.; Mee, E.T.; Collot-Teixeira, S.; Anderson, R.; Schepelmann, S.; Minor, P.D.; Delwart, E. Comparing viral metagenomics methods using a highly multiplexed human viral pathogens reagent. J. Virol. Methods 2015, 213, 139–146. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdóttir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Pichon, M.; Picard, C.; Casalegno, J.S.; Valette, M.; Schuffenecker, I.; Billard, L.; Vallet, S.; Vilchez, G.; Cheynet, V.; et al. Quality control implementation for universal characterization of DNA and RNA viruses in clinical respiratory samples using single metagenomic next-generation sequencing workflow. Bmc Infect. Dis. 2018, 18, 537. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Chessa, C.; Bourgoin, A.; Giraudeau, G.; Plouzeau, C.; Agius, G.; Lévêque, N.; Beby-Defaux, A. Comparison of eMAGTM versus NucliSENS® EasyMAG® performance on clinical specimens. J. Clin. Virol. 2017, 88, 52–57. [Google Scholar] [CrossRef]
- Plyusnin, I.; Kant, R.; Jääskeläinen, A.J.; Sironen, T.; Holm, L.; Vapalahti, O.; Smura, T. Novel NGS Pipeline for Virus Discovery from a Wide Spectrum of Hosts and Sample Types. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Rose, J.A.; Hoggan, M.D.; Shatkin, A.J. Nucleic acid from an adeno-associated virus: Chemical and physical studies. Proc. Natl. Acad. Sci. USA 1966, 56, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Kwe Yinda, K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef]
- Gagnieur, L.; Cheval, J.; Gratigny, M.; Hébert, C.; Muth, E.; Dumarest, M.; Eloit, M. Unbiased analysis by high throughput sequencing of the viral diversity in fetal bovine serum and trypsin used in cell culture. Biologicals 2014, 42, 145–152. [Google Scholar] [CrossRef]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.-J.; et al. Virome of US bovine calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Virus | Virus Family | Molecular Typing | Baltimore Classification | Genome Composition | Genome Size (kb) | Virion Size (nm) | Enveloped | Ct Value |
|---|---|---|---|---|---|---|---|---|---|
| Adenovirus | Adenoviridae | ADV-A31 | Group I: dsDNA | Linear | 34 | 65/80 | No | 21.1 | |
| Respiratory Syncytial Virus | Paramyxoviridae | RSV-A | Group V: ssRNA (-) | Linear | 15 | 150 | Yes | 22.1 | |
| Mock Virome | Herpes Simplex Virus | Herpesviridae | HSV-1 | Group I: dsDNA | Linear | 150 | 120/300 | Yes | 26.2 |
| Influenza Virus | Orthomyxoviridae | IAV | Group V: ssRNA (-) | Segmented | 13 | 80/120 | Yes | 20.4 | |
| Rhinovirus | Picornaviridae | HRV-A13 | Group IV: ssRNA (+) | Linear | 7 | 30 | No | 29 | |
| Clinical samples | Herpes Simplex Virus | Herpesviridae | HSV-1 | Group I: dsDNA | Linear | 150 | 120/300 | Yes | 16.6 |
| Respiratory Syncytial Virus | Paramyxoviridae | RSV | Group V: ssRNA (-) | Linear | 15.2 | 150 | Yes | 19.3 |
| Virus | Cells | Nature of the Sample | Culture Media | Additional Elements | Number of Days in Culture | Cryoprotectant Medium |
|---|---|---|---|---|---|---|
| Adenovirus | Hep | stool | MEM | 2% penicillin-streptomycin + 1% L-glutamine + 0.05% neomycin + 2% FBS + 2% Hepes Buffer | 4 | Yes |
| Respiratory Syncytial Virus | Hep | nasal throat | MEM | 2% penicillin-streptomycin + 1% L-glutamine + 0.05% neomycin + 2% FBS + 2% Hepes Buffer | 4 | Yes |
| Herpes Simplex Virus | Vero | vaginal swab | MEM199 | 2% penicillin-streptomycin | 3 | No |
| Influenza A | MDCK | bronchoalveolar lavage | MEM | 2% penicillin-streptomycin + 1% L-glutamine + 0.05% neomycin + 0.05% Trypsine + 2% Hepes Buffer | 5 | Yes |
| Rhinovirus | MRC5 | tracheobronchial aspiration | MEM | 2% penicillin-streptomycin + 1% L-glutamine + 0.05% neomycin + 2% FBS + 2% Hepes Buffer | 5 | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabatier, M.; Bal, A.; Destras, G.; Regue, H.; Quéromès, G.; Cheynet, V.; Lina, B.; Bardel, C.; Brengel-Pesce, K.; Navratil, V.; et al. Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses. Microorganisms 2020, 8, 1539. https://doi.org/10.3390/microorganisms8101539
Sabatier M, Bal A, Destras G, Regue H, Quéromès G, Cheynet V, Lina B, Bardel C, Brengel-Pesce K, Navratil V, et al. Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses. Microorganisms. 2020; 8(10):1539. https://doi.org/10.3390/microorganisms8101539
Chicago/Turabian StyleSabatier, Marina, Antonin Bal, Grégory Destras, Hadrien Regue, Grégory Quéromès, Valérie Cheynet, Bruno Lina, Claire Bardel, Karen Brengel-Pesce, Vincent Navratil, and et al. 2020. "Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses" Microorganisms 8, no. 10: 1539. https://doi.org/10.3390/microorganisms8101539
APA StyleSabatier, M., Bal, A., Destras, G., Regue, H., Quéromès, G., Cheynet, V., Lina, B., Bardel, C., Brengel-Pesce, K., Navratil, V., & Josset, L. (2020). Comparison of Nucleic Acid Extraction Methods for a Viral Metagenomics Analysis of Respiratory Viruses. Microorganisms, 8(10), 1539. https://doi.org/10.3390/microorganisms8101539