Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool

 , ,
, ,  and
and
Abstract
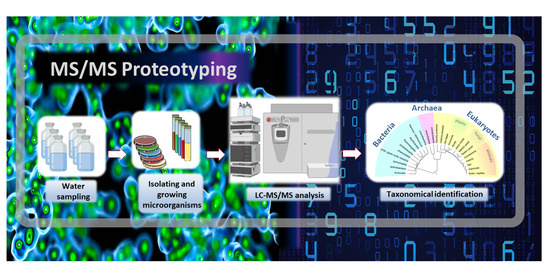
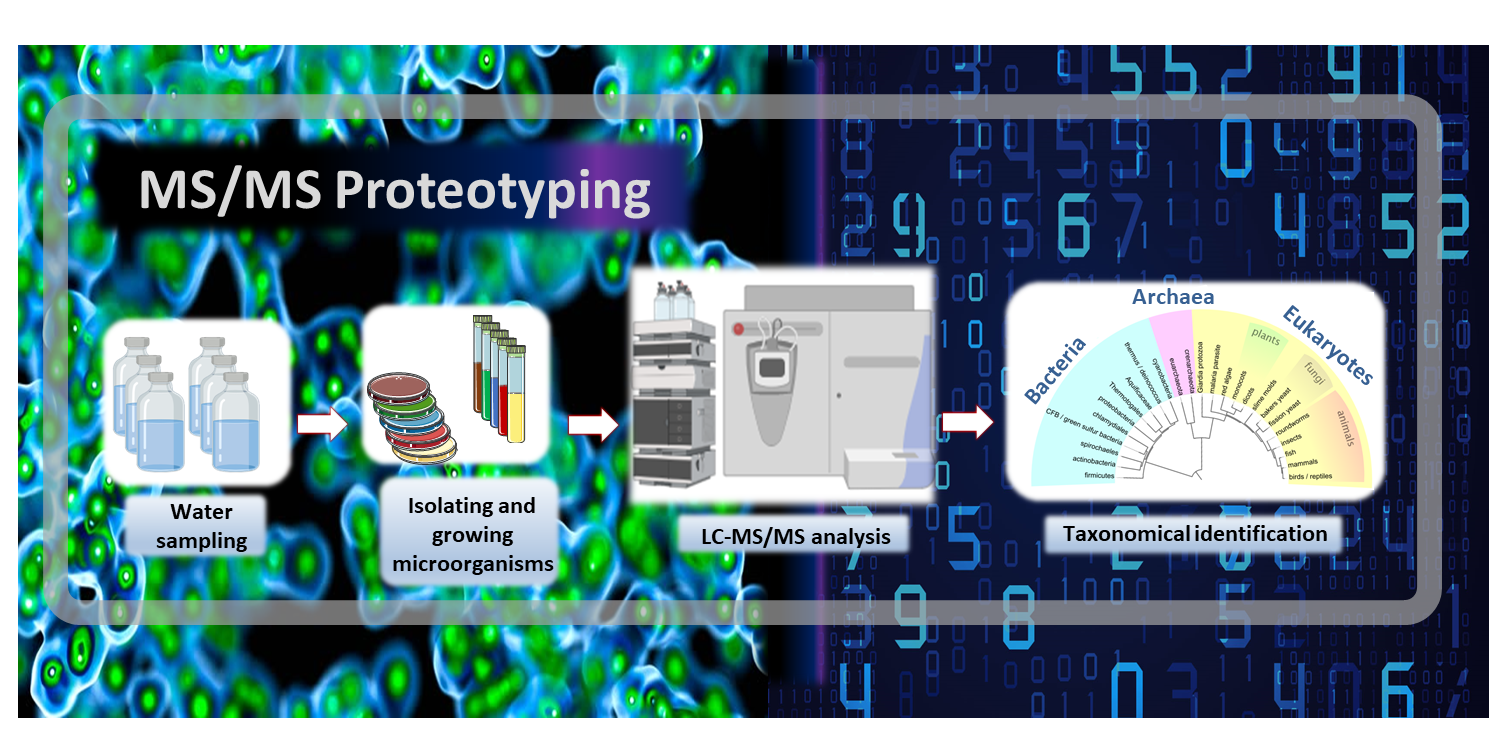
1. Introduction
2. Materials and Methods
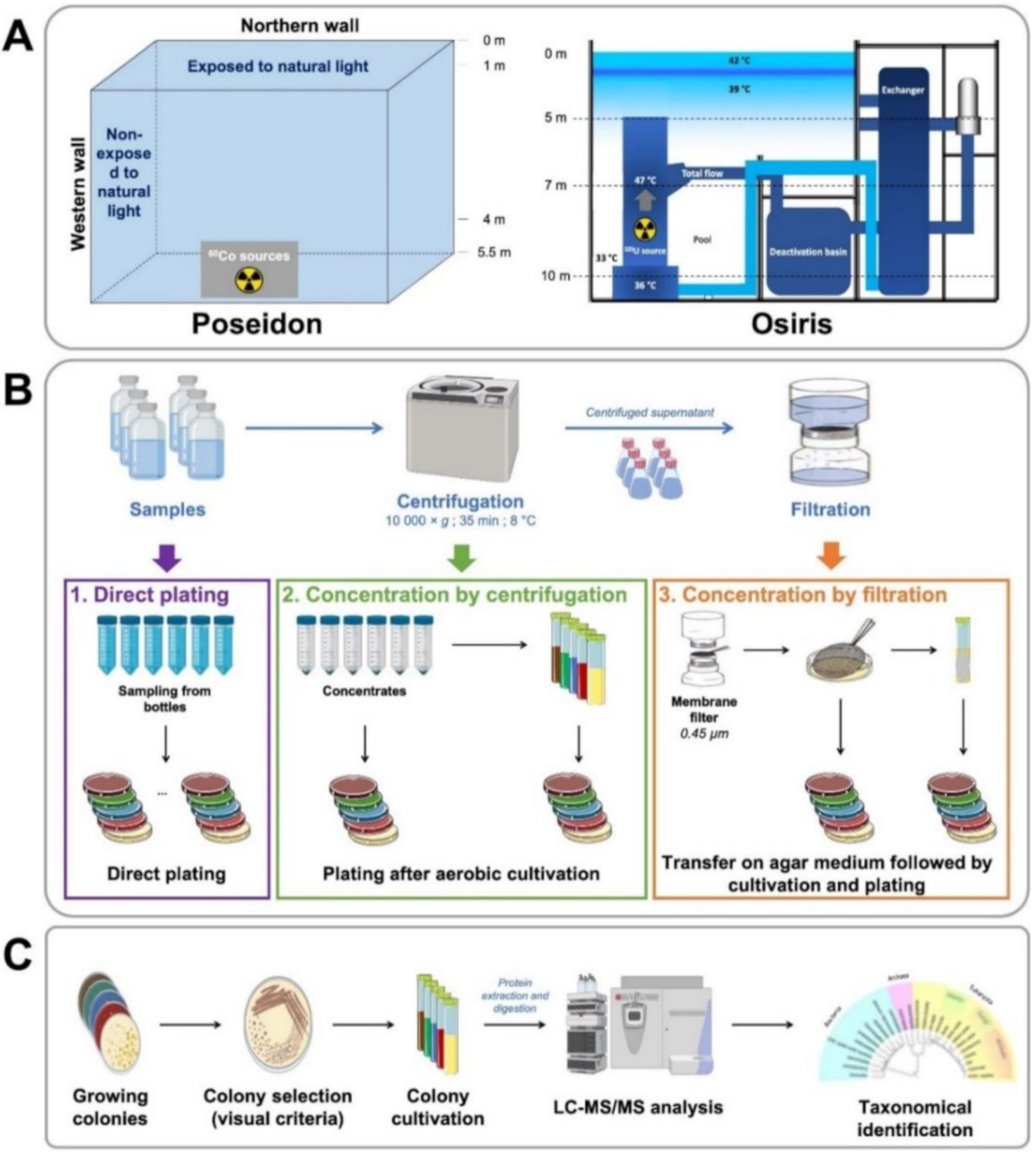
2.1. Microorganism Sampling
2.2. Culture and Isolation
2.3. Proteotyping of Isolates by Tandem Mass Spectrometry
2.4. Proteotyping Data
3. Results
3.1. Sampling and Culture Strategies to Obtain Diverse Isolates
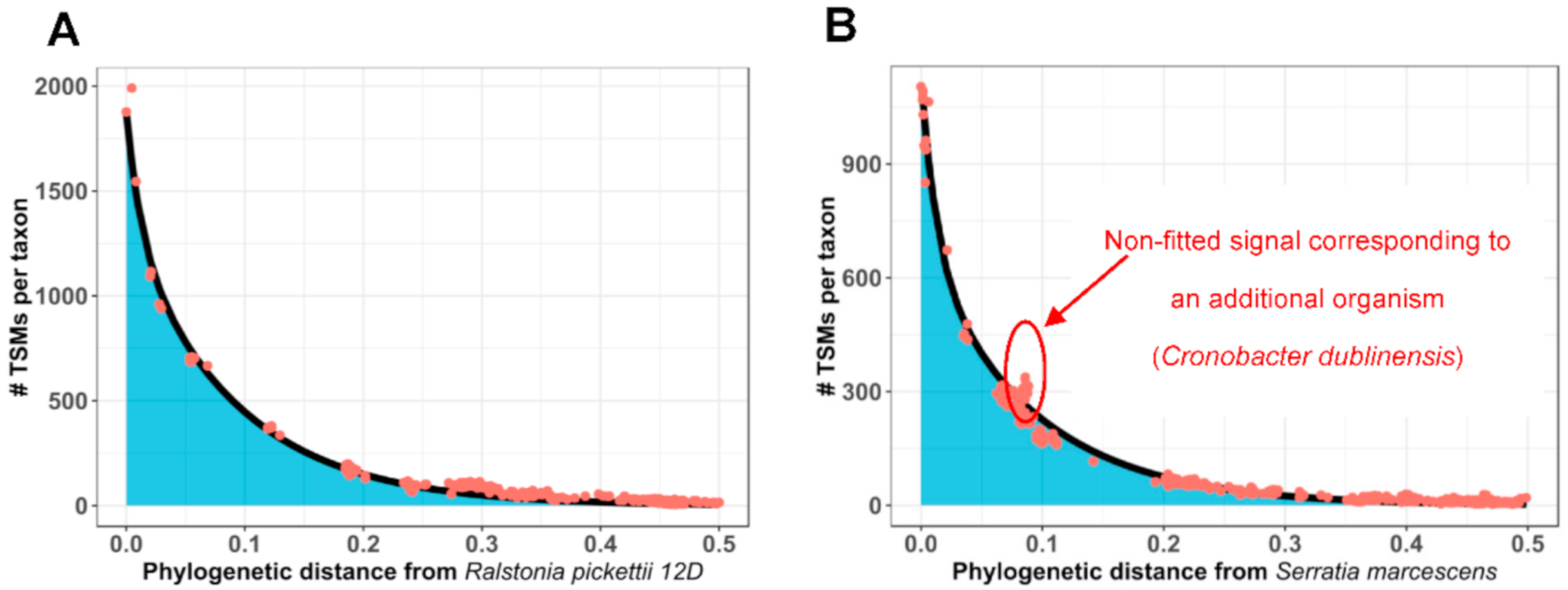
3.2. How Phylopeptidomics Can Be Used to Verify Whether a Sample is Mono-Organism or Contains a Mixture of Microorganisms
3.3. Examples of Species-Level Identification and Biomass Quantification for Mixtures of Microorganisms
3.4. Phylopeptidomics also Successfully Identifies Fungi
3.5. Analysis of the Whole Cohort of Microorganisms Identified
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Togawa, Y.; Shiotani, S.; Kato, Y.; Ezaki, K.; Nunoshiba, T.; Hiratsu, K. Development of a supF-based mutation-detection system in the extreme thermophile Thermus thermophilus HB27. Mol. Genet. Genom. 2019, 294, 1085–1093. [Google Scholar] [CrossRef]
- Rubiano-Labrador, C.; Bland, C.; Miotello, G.; Guerin, P.; Pible, O.; Baena, S.; Armengaud, J. Proteogenomic insights into salt tolerance by a halotolerant alpha-proteobacterium isolated from an Andean saline spring. J. Proteom. 2014, 97, 36–47. [Google Scholar] [CrossRef]
- Zivanovic, Y.; Armengaud, J.; Lagorce, A.; Leplat, C.; Guerin, P.; Dutertre, M.; Anthouard, V.; Forterre, P.; Wincker, P.; Confalonieri, F. Genome analysis and genome-wide proteomics of Thermococcus gammatolerans, the most radioresistant organism known amongst the Archaea. Genome Biol. 2009, 10, R70. [Google Scholar] [CrossRef]
- De Groot, A.; Dulermo, R.; Ortet, P.; Blanchard, L.; Guerin, P.; Fernandez, B.; Vacherie, B.; Dossat, C.; Jolivet, E.; Siguier, P.; et al. Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet. 2009, 5, e1000434. [Google Scholar] [CrossRef] [PubMed]
- McKenney, P.T.; Driks, A.; Eichenberger, P. The Bacillus subtilis endospore: Assembly and functions of the multilayered coat. Nat. Rev. Microbiol. 2013, 11, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001. [Google Scholar] [CrossRef] [PubMed]
- Hermon, L.; Denonfoux, J.; Hellal, J.; Joulian, C.; Ferreira, S.; Vuilleumier, S.; Imfeld, G. Dichloromethane biodegradation in multi-contaminated groundwater: Insights from biomolecular and compound-specific isotope analyses. Water Res. 2018, 142, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J. Next-generation proteomics faces new challenges in environmental biotechnology. Curr. Opin. Biotechnol. 2016, 38, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Kruger, A.; Schafers, C.; Schroder, C.; Antranikian, G. Towards a sustainable biobased industry-Highlighting the impact of extremophiles. New Biotechnol. 2018, 40 Pt A, 144–153. [Google Scholar] [CrossRef]
- Lagier, J.C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.; Albertsen, M. Investigation of Detection Limits and the Influence of DNA Extraction and Primer Choice on the Observed Microbial Communities in Drinking Water Samples Using 16S rRNA Gene Amplicon Sequencing. Front. Microbiol. 2018, 9, 2140. [Google Scholar] [CrossRef] [PubMed]
- Rutanga, J.P.; Van Puyvelde, S.; Heroes, A.S.; Muvunyi, C.M.; Jacobs, J.; Deborggraeve, S. 16S metagenomics for diagnosis of bloodstream infections: Opportunities and pitfalls. Expert Rev. Mol. Diagn. 2018, 18, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Grenga, L.; Pible, O.; Armengaud, J. Pathogen proteotyping: A rapidly developing application of mass spectrometry to address clinical concerns. Clin. Mass Spectrom. 2019, 14, 9–17. [Google Scholar] [CrossRef]
- Karlsson, R.; Gonzales-Siles, L.; Gomila, M.; Busquets, A.; Salva-Serra, F.; Jaen-Luchoro, D.; Jakobsson, H.E.; Karlsson, A.; Boulund, F.; Kristiansson, E.; et al. Proteotyping bacteria: Characterization, differentiation and identification of pneumococcus and other species within the Mitis Group of the genus Streptococcus by tandem mass spectrometry proteomics. PLoS ONE 2018, 13, e0208804. [Google Scholar] [CrossRef]
- Hayoun, K.; Gaillard, J.C.; Pible, O.; Alpha-Bazin, B.; Armengaud, J. High-throughput proteotyping of bacterial isolates by double barrel chromatography-tandem mass spectrometry based on microplate paramagnetic beads and phylopeptidomics. J. Proteom. 2020, 226, 103887. [Google Scholar] [CrossRef]
- Mesuere, B.; Willems, T.; Van der Jeugt, F.; Devreese, B.; Vandamme, P.; Dawyndt, P. Unipept web services for metaproteomics analysis. Bioinformatics 2016, 32, 1746–1748. [Google Scholar] [CrossRef]
- Padliya, N.D.; Garrett, W.M.; Campbell, K.B.; Tabb, D.L.; Cooper, B. Tandem mass spectrometry for the detection of plant pathogenic fungi and the effects of database composition on protein inferences. Proteomics 2007, 7, 3932–3942. [Google Scholar] [CrossRef]
- Verberkmoes, N.C.; Hervey, W.J.; Shah, M.; Land, M.; Hauser, L.; Larimer, F.W.; Van Berkel, G.J.; Goeringer, D.E. Evaluation of “shotgun” proteomics for identification of biological threat agents in complex environmental matrixes: Experimental simulations. Anal. Chem. 2005, 77, 923–932. [Google Scholar] [CrossRef]
- Pible, O.; Allain, F.; Jouffret, V.; Culotta, K.; Miotello, G.; Armengaud, J. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics”. Microbiome 2020, 8, 30. [Google Scholar] [CrossRef]
- Hayoun, K.; Gouveia, D.; Grenga, L.; Pible, O.; Armengaud, J.; Alpha-Bazin, B. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 2019, 10, 1985. [Google Scholar] [CrossRef]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 1985, 49, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mappa, C.; Pible, O.; Armengaud, J.; Alpha-Bazin, B. Assessing the ratio of Bacillus spores and vegetative cells by shotgun proteomics. Environ. Sci. Pollut. Res. Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, E.M.; Allain, F.; Gaillard, J.C.; Pible, O.; Armengaud, J. Taking the shortcut for high-throughput shotgun proteomic analysis of bacteria. Methods Mol. Biol. 2014, 1197, 275–285. [Google Scholar]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Kepler, R.M.; Luangsa-Ard, J.J.; Hywel-Jones, N.L.; Quandt, C.A.; Sung, G.H.; Rehner, S.A.; Aime, M.C.; Henkel, T.W.; Sanjuan, T.; Zare, R.; et al. A phylogenetically-based nomenclature for Cordycipitaceae (Hypocreales). IMA Fungus 2017, 8, 335–353. [Google Scholar] [CrossRef]
- Bernard, G.; Pathmanathan, J.S.; Lannes, R.; Lopez, P.; Bapteste, E. Microbial Dark Matter Investigations: How Microbial Studies Transform Biological Knowledge and Empirically Sketch a Logic of Scientific Discovery. Genome Biol. Evol. 2018, 10, 707–715. [Google Scholar] [CrossRef]
- Saw, J.H.; Spang, A.; Zaremba-Niedzwiedzka, K.; Juzokaite, L.; Dodsworth, J.A.; Murugapiran, S.K.; Colman, D.R.; Takacs-Vesbach, C.; Hedlund, B.P.; Guy, L.; et al. Exploring microbial dark matter to resolve the deep archaeal ancestry of eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140328. [Google Scholar] [CrossRef]
- Sheu, S.Y.; Chen, W.T.; Young, C.C.; Chen, W.M. Rheinheimera coerulea sp. nov. isolated from a freshwater creek, and emended description of genus Rheinheimera Brettar et al. 2002. Int. J. Syst. Evol. Microbiol. 2018, 68, 2340–2347. [Google Scholar] [CrossRef]
- Pible, O.; Armengaud, J. Improving the quality of genome, protein sequence, and taxonomy databases: A prerequisite for microbiome meta-omics 2.0. Proteomics 2015, 15, 3418–3423. [Google Scholar] [CrossRef]
- Murray, A.E.; Freudenstein, J.; Gribaldo, S.; Hatzenpichler, R.; Hugenholtz, P.; Kampfer, P.; Konstantinidis, K.T.; Lane, C.E.; Papke, R.T.; Parks, D.H.; et al. Roadmap for naming uncultivated Archaea and Bacteria. Nat. Microbiol. 2020, 5, 987–994. [Google Scholar] [CrossRef]
- Connon, S.A.; Giovannoni, S.J. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef] [PubMed]
- Chicote, E.; Moreno, D.A.; Garcia, A.M.; Sarro, M.I.; Lorenzo, P.I.; Montero, F. Biofouling on the walls of a spent nuclear fuel pool with radioactive ultrapure water. Biofouling 2004, 20, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Sarro, M.I.; Garcia, A.M.; Moreno, D.A.; Montero, F. Development and characterization of biofilms on stainless steel and titanium in spent nuclear fuel pools. J. Ind. Microbiol. Biotechnol. 2007, 34, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Rivasseau, C.; Farhi, E.; Compagnon, E.; de Gouvion Saint Cyr, D.; van Lis, R.; Falconet, D.; Kuntz, M.; Atteia, A.; Coute, A. Coccomyxa actinabiotis sp. nov. (Trebouxiophyceae, Chlorophyta), a new green microalga living in the spent fuel cooling pool of a nuclear reactor. J. Phycol. 2016, 52, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Gallois, N.; Alpha-Bazin, B.; Ortet, P.; Barakat, M.; Piette, L.; Long, J.; Berthomieu, C.; Armengaud, J.; Chapon, V. Proteogenomic insights into uranium tolerance of a Chernobyl’s Microbacterium bacterial isolate. J. Proteom. 2018, 177, 148–157. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
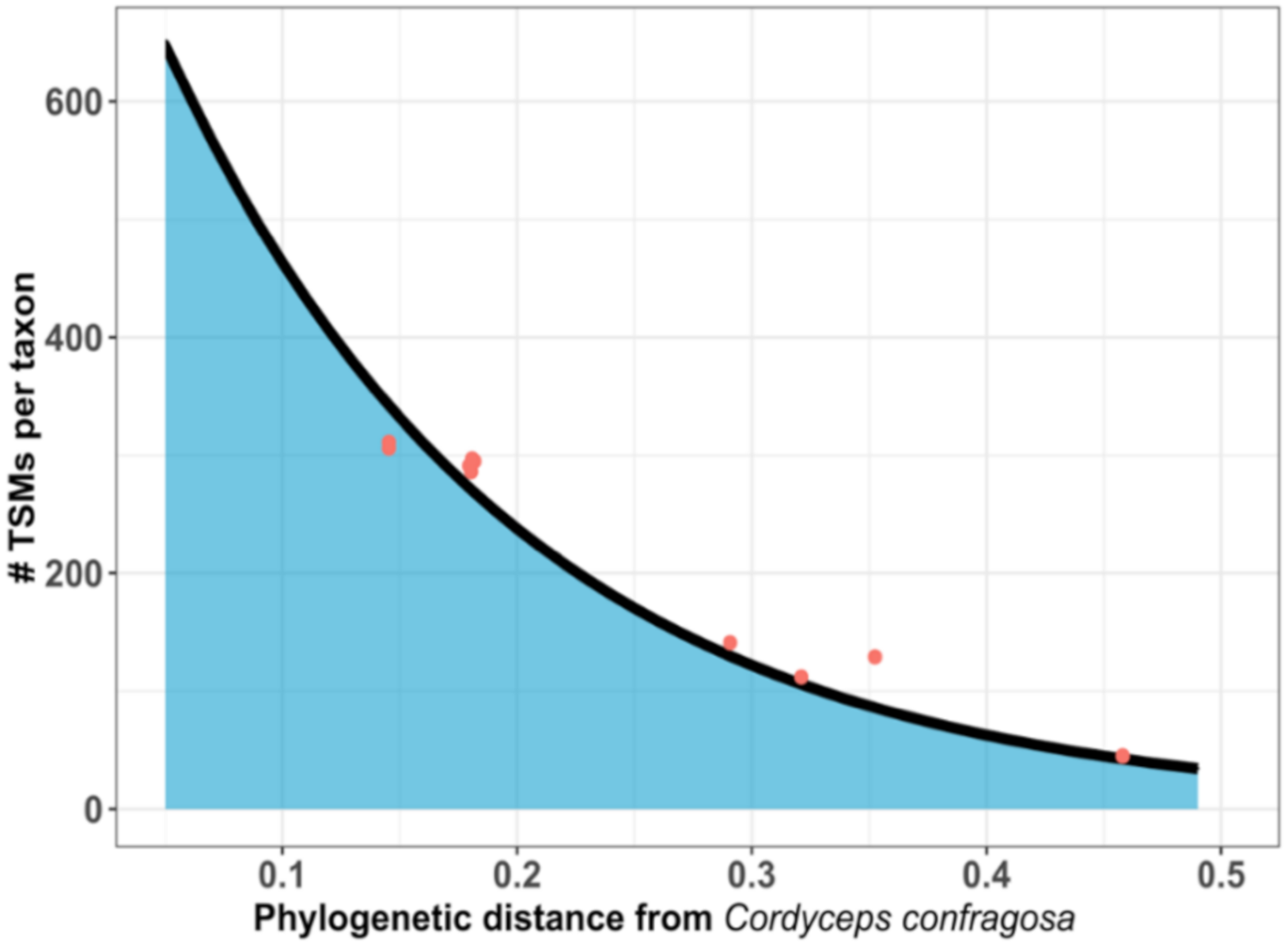{kind=link}
| Sample | MS/MS Spectra | Total PSMs | Organism(s) | Tax ID | Species-Assigned TSMs | Species-Specific Peptides | Genus-Specific Peptides |
|---|---|---|---|---|---|---|---|
| A | 3988 | 2020 | Ralstonia pickettii | 329 | 1990 | 130 | 338 |
| B | 3293 | 750 | Nevskia ramosa | 64002 | 430 | 143 | 158 |
| Limnobacter thiooxidans | 131080 | 150 | 73 | 73 | |||
| C | 2740 | 907 | Cordyceps confragosa | 1105325 | 685 | 44 | 74 |
| D | 2951 | 576 | Bacillus cereus | 1396 | 514 | 2 | 33 |
| E | 3001 | 747 | Bacillus indicus | 246786 | 580 | 2 | 174 |
| F | 3369 | 1210 | Bacillus thuringiensis | 1428 | 1195 | 43 | 221 |
| G | 3223 | 738 | Bosea vaviloviae | 1526658 | 465 | 25 | 246 |
| H | 4135 | 638 | Brevundimonas bacteroides | 74311 | 436 | 29 | 275 |
| I | 1955 | 364 | Hymenobacter swuensis | 1446467 | 296 | 48 | 189 |
| J | 4264 | 943 | Limnobacter thiooxidans | 131080 | 861 | 418 | 418 |
| K | 3765 | 1350 | Massilia timonae | 47229 | 1283 | 516 | 656 |
| L | 427 | 106 | Methylobacterium extorquens | 408 | 67 | 3 | 26 |
| M | 3356 | 846 | Microbacterium oxydans | 82380 | 771 | 47 | 466 |
| N | 3407 | 460 | Nevskia ramosa | 64002 | 355 | 123 | 140 |
| O | 3007 | 868 | Pantoea vagans | 470934 | 758 | 13 | 102 |
| P | 3871 | 621 | Porphyrobacter cryptus | 196588 | 410 | 38 | 162 |
| Q | 3479 | 1259 | Propionibacterium acnes | 1747 | 1228 | 15 | 585 |
| R | 4142 | 1111 | Sphingomonas echinoides | 59803 | 1009 | 231 | 607 |
| S | 4139 | 835 | Sphingomonas elodea | 179878 | 668 | 97 | 362 |
| T | 3619 | 1214 | Sphingomonas parapaucimobilis | 28213 | 934 | 180 | 713 |
| U | 1352 | 262 | Xanthobacter autotrophicus | 280 | 144 | 13 | 29 |
| V | 1509 | 153 | Cladosporium herbarum | 29918 | 12 | 7 | 7 |
| W | 2209 | 316 | Limnobacter thiooxidans | 131080 | 94 | 55 | 55 |
| Bacillus | 1386 | 133 | / | 40 | |||
| Hymenobacter norwichensis | 223903 | 40 | 20 | 31 | |||
| X | 2054 | 496 | Rhodococcus erythropolis | 1833 | 338 | 4 | 179 |
| Massilia timonae | 47229 | 97 | 49 | 61 | |||
| Y | 3451 | 661 | Sphingomonas parapaucimobilis | 28213 | 182 | 10 | 129 |
| Caulobacter vibrioides | 155892 | 160 | 33 | 72 | |||
| Limnobacter thiooxidans | 391597 | 113 | 54 | 54 | |||
| Z | 3972 | 454 | Variovorax paradoxus | 34073 | 194 | 2 | 7 |
| Acidovorax | 12916 | 95 | / | 5 | |||
| Polaromonas | 52972 | 33 | / | 6 | |||
| AA | 3760 | 516 | Rheinheimera texasensis | 306205 | 324 | 35 | 87 |
| Limnobacter thiooxidans | 131080 | 37 | 23 | 23 | |||
| AB | 1840 | 356 | Cellulomonas gilvus | 11 | 86 | 1 | 23 |
| Sphingomonas parapaucimobilis | 28213 | 137 | 11 | 77 | |||
| AC | 4349 | 623 | Xanthobacter autotrophicus | 280 | 357 | 23 | 70 |
| Azorhizobium caulinodans | 7 | 52 | 3 | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayoun, K.; Pible, O.; Petit, P.; Allain, F.; Jouffret, V.; Culotta, K.; Rivasseau, C.; Armengaud, J.; Alpha-Bazin, B. Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool. Microorganisms 2020, 8, 1525. https://doi.org/10.3390/microorganisms8101525
Hayoun K, Pible O, Petit P, Allain F, Jouffret V, Culotta K, Rivasseau C, Armengaud J, Alpha-Bazin B. Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool. Microorganisms. 2020; 8(10):1525. https://doi.org/10.3390/microorganisms8101525
Chicago/Turabian StyleHayoun, Karim, Olivier Pible, Pauline Petit, François Allain, Virginie Jouffret, Karen Culotta, Corinne Rivasseau, Jean Armengaud, and Béatrice Alpha-Bazin. 2020. "Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool" Microorganisms 8, no. 10: 1525. https://doi.org/10.3390/microorganisms8101525
APA StyleHayoun, K., Pible, O., Petit, P., Allain, F., Jouffret, V., Culotta, K., Rivasseau, C., Armengaud, J., & Alpha-Bazin, B. (2020). Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool. Microorganisms, 8(10), 1525. https://doi.org/10.3390/microorganisms8101525