Temporal Changes in Patient-Matched Staphylococcus epidermidis Isolates from Infections: towards Defining a ‘True’ Persistent Infection

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolate Collection and Genomes
2.2. Reference Pan-Genome
2.3. Analysis of Persistent Infection Isolates
2.4. Multilocus Sequence Typing (MLST)
2.5. Statistical Analyses
3. Results
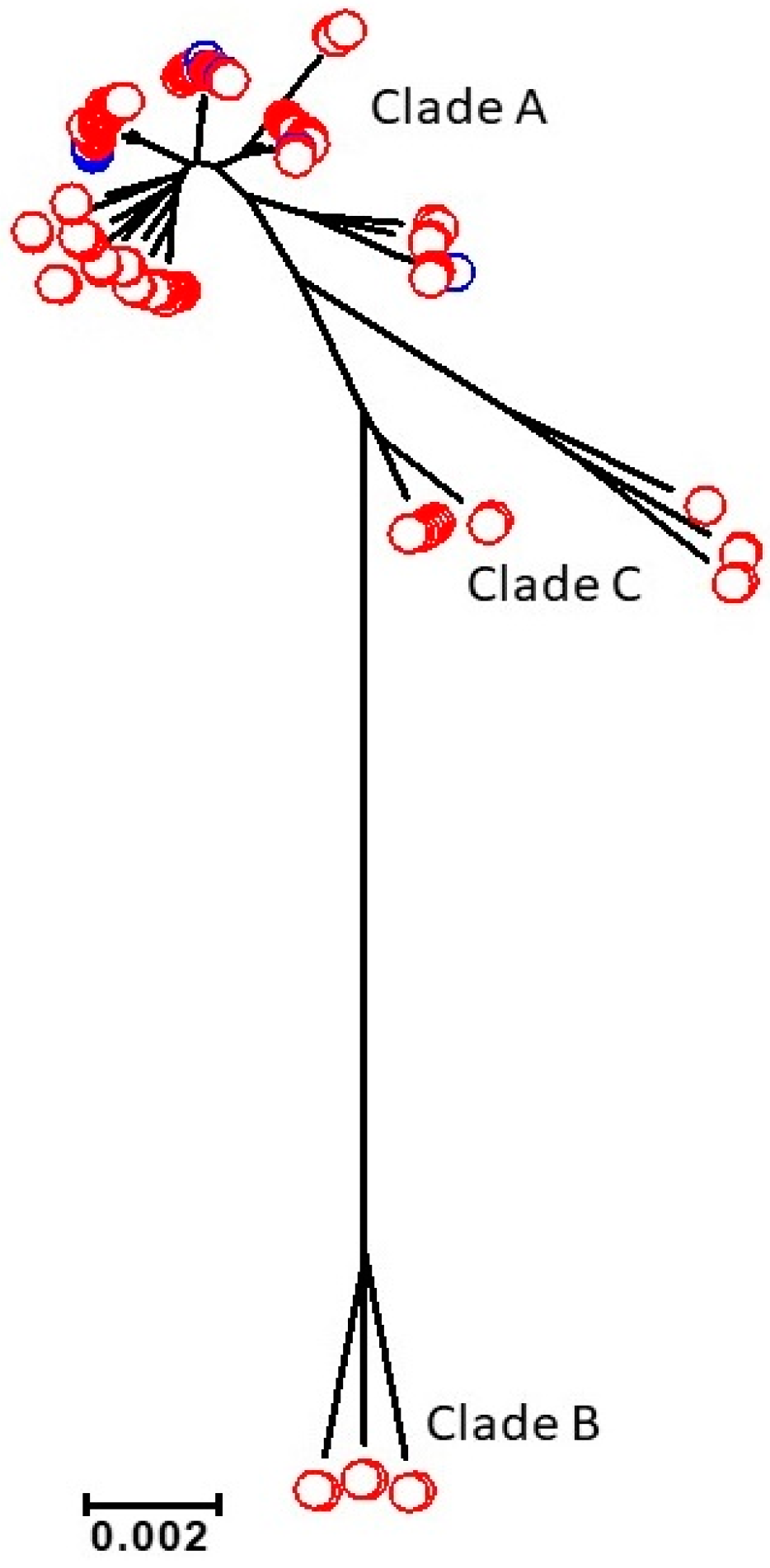
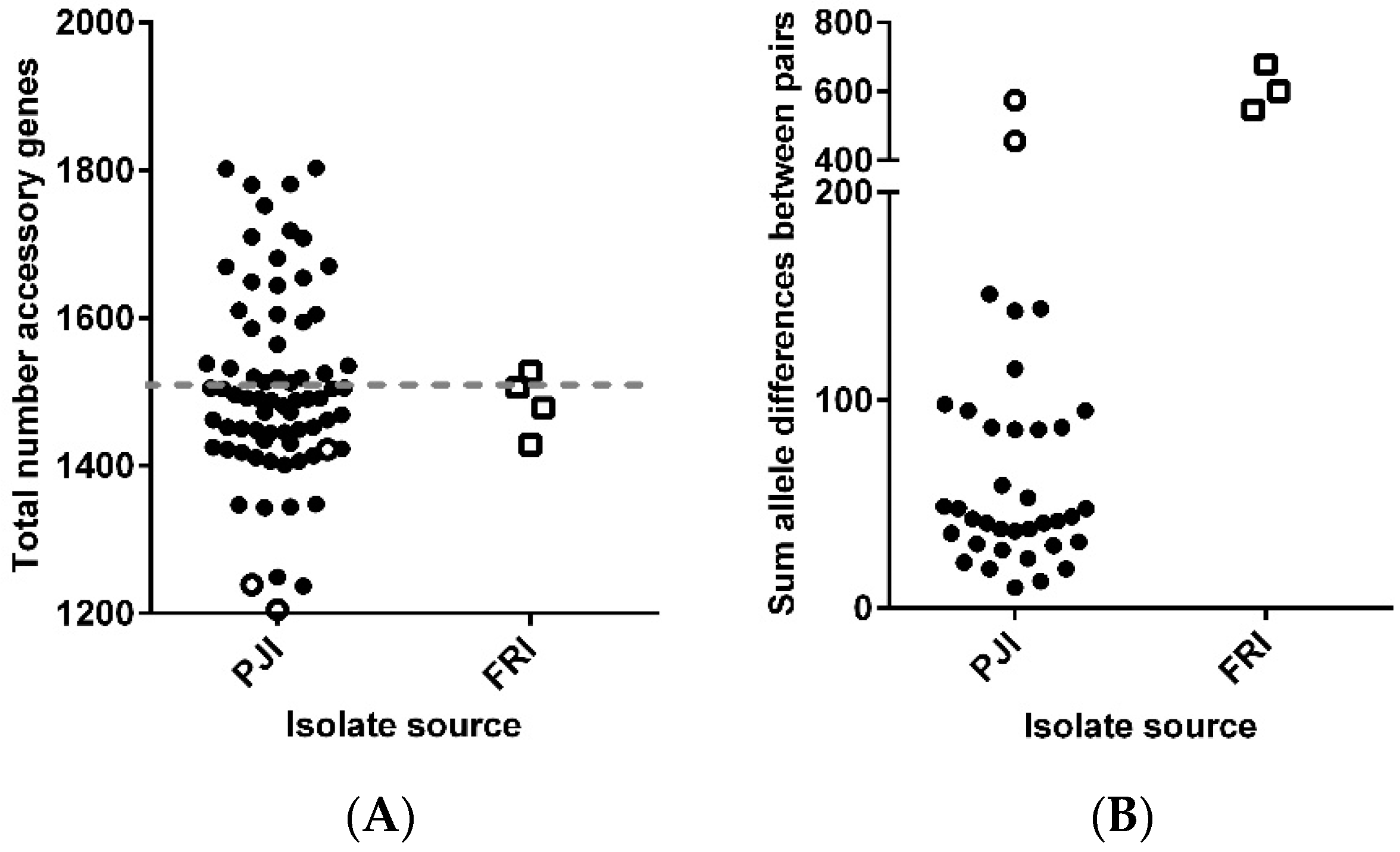
3.1. Descriptive Genomics for 115 Persistent Infection Isolates
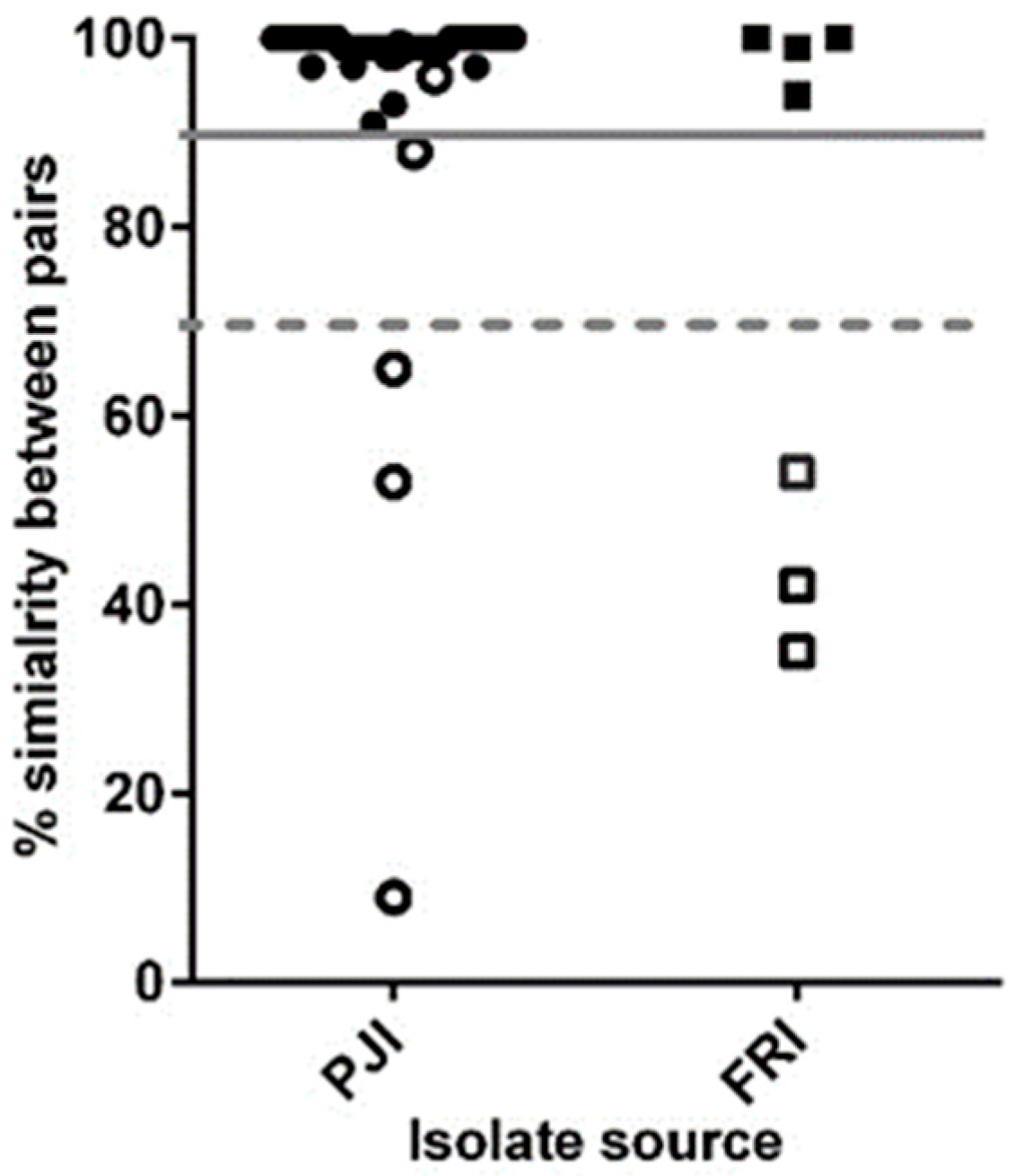
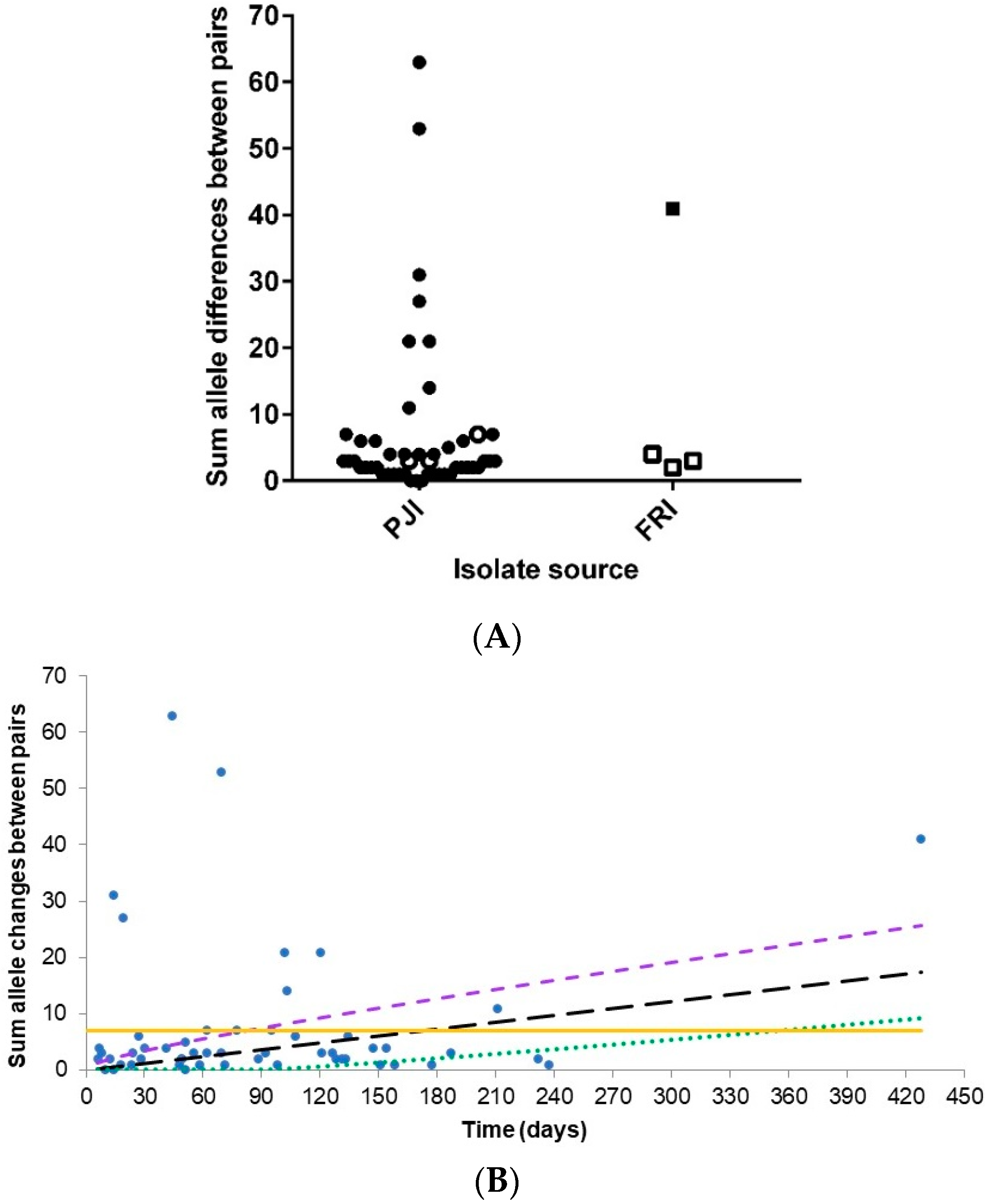
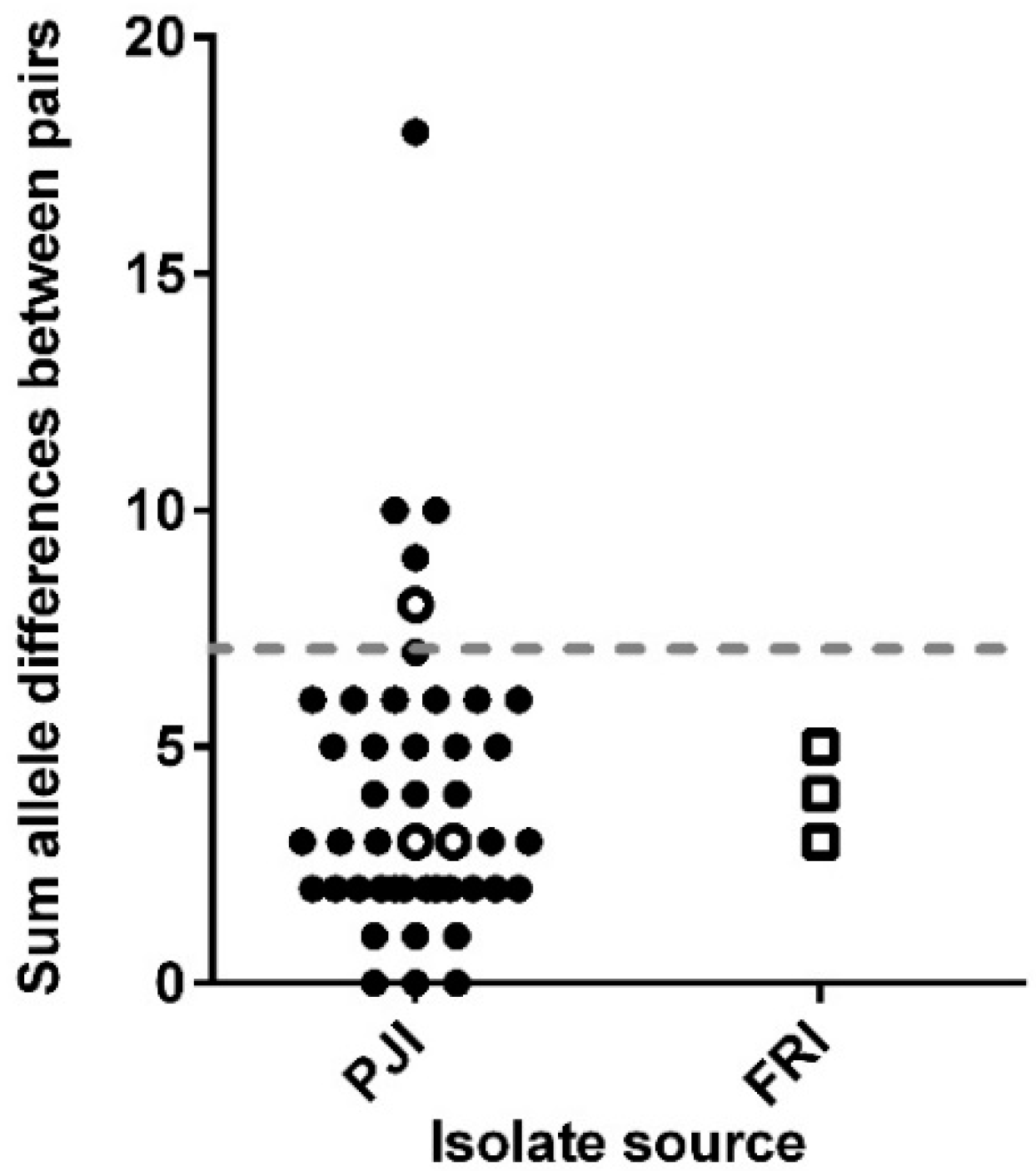
3.2. Core Genome Analysis for 102 Persistent Infection Isolates (49 Patients)
3.3. Analysis of the Core and Accessory Genomes of ‘True’ Persistent Infection Isolates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Piette, A.; Verschraegen, G. Role of coagulase-negative staphylococci in human disease. Vet. Microbiol. 2009, 134, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sabate Bresco, M.; Harris, L.G.; Thompson, K.; Stanic, B.; Morgenstern, M.; O’Mahony, L.; Richards, R.G.; Moriarty, T.F. Pathogenic mechanisms and host interactions in Staphylococcus epidermidis device-related infection. Front. Microbiol. 2017, 8, 1401. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Murray, S.; Pascoe, B.; Bray, J.; Meric, G.; Magerios, L.; Wilkinson, T.S.; Jeeves, R.; Rohde, H.; Schwarz, S.; et al. Biofilm morphotypes and population structure among Staphylococcus epidermidis from commensal and clinical samples. PLoS ONE 2016, 11, e0151240. [Google Scholar] [CrossRef]
- Espadinha, D.; Sobral, R.G.; Mendes, C.I.; Meric, G.; Sheppard, S.K.; Carrico, J.A.; de Lencastre, H.; Miragaia, M. Distinct phenotypic and genomic signatures underlie contrasting pathogenic potential of Staphylococcus epidermidis clonal lineages. Front. Microbiol. 2019, 10, 1971. [Google Scholar] [CrossRef]
- Mack, D.; Horstkotte, M.; Rohde, H.; Knobloch, J. Coagulase-Negative Staphylococci; CRC Press: Boca Ralton, FL, USA, 2006; pp. 109–153. [Google Scholar]
- Young, B.C.; Golubchik, T.; Batty, E.M.; Fung, R.; Larner-Svensson, H.; Votintseva, A.A.; Miller, R.R.; Godwin, H.; Knox, K.; Everitt, R.G.; et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc. Natl. Acad. Sci. USA 2012, 109, 4550–4555. [Google Scholar] [CrossRef]
- Golubchik, T.; Batty, E.M.; Miller, R.R.; Farr, H.; Young, B.C.; Larner-Svensson, H.; Fung, R.; Godwin, H.; Knox, K.; Votintseva, A.; et al. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS ONE 2013, 8, e61319. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Ferry, T.; Uckay, I.; Vaudaux, P.; Francois, P.; Schrenzel, J.; Harbarth, S.; Laurent, F.; Bernard, L.; Vandenesch, F.; Etienne, J.; et al. Risk factors for treatment failure in orthopedic device-related methicillin-resistant Staphylococcus aureus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 171–180. [Google Scholar] [CrossRef]
- Rohde, H.; Burandt, E.C.; Siemssen, N.; Frommelt, L.; Burdelski, C.; Wurster, S.; Scherpe, S.; Davies, A.P.; Harris, L.G.; Horstkotte, M.A.; et al. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials 2007, 28, 1711–1720. [Google Scholar] [CrossRef]
- Morgenstern, M.; Post, V.; Erichsen, C.; Hungerer, S.; Buhren, V.; Militz, M.; Richards, G.; Moriarty, F. Biofilm formation increases treatment failure in Staphylococcus epidermidis device-related osteomyelitis of the lower extremity in human patients. J. Orthop. Res. 2016. [Google Scholar] [CrossRef]
- Post, V.; Harris, L.G.; Morgenstern, M.; Mageiros, L.; Hitchings, M.D.; Meric, G.; Pascoe, B.; Sheppard, S.K.; Richards, R.G.; Moriarty, T.F. Comparative genomics study of Staphylococcus epidermidis isolates from orthopedic-device-related infections correlated with patient outcome. J. Clin. Microbiol. 2017, 55, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; DeBoy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar] [CrossRef]
- Zhang, Y.-Q.; Ren, S.-X.; Li, H.-L.; Wang, Y.-X.; Fu, G.; Yang, J.; Qin, Z.-Q.; Miao, Y.-G.; Wang, W.-Y.; Chen, R.-S.; et al. Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol. Microbiol. 2003, 49, 1577–1593. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Thomas, J.C.; Vargas, M.R.; Miragaia, M.; Peacock, S.J.; Archer, G.L.; Enright, M.C. Improved Multilocus Sequence Typing scheme for Staphylococcus epidermidis. J. Clin. Microbiol. 2007, 45, 616–619. [Google Scholar] [CrossRef]
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef]
- Van Eldere, J.; Peetermans, W.E.; Struelens, M.; Deplano, A.; Bobbaers, H. Polyclonal Staphylococcal endocarditis caused by genetic variability. Clin. Infect. Dis. 2000, 31, 24–30. [Google Scholar] [CrossRef]
- Gomez-Lunar, Z.; Hernandez-Gonzalez, I.; Rodriguez-Torres, M.D.; Souza, V.; Olmedo-Alvarez, G. Microevolution Analysis of Bacillus coahuilensis Unveils Differences in Phosphorus Acquisition Strategies and Their Regulation. Front. Microbiol. 2016, 7, 58. [Google Scholar] [CrossRef]
- Meric, G.; Miragaia, M.; de Been, M.; Yahara, K.; Pascoe, B.; Mageiros, L.; Mikhail, J.; Harris, L.G.; Wilkinson, T.S.; Rolo, J.; et al. Ecological Overlap and Horizontal Gene Transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 2015, 7, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Galdbart, J.-O.; Morvan, A.; Desplaces, N.; el Solh, N. Phenotypic and genomic variation among Staphylococcus epidermidis strains infecting joint prostheses. J. Clin. Microbiol. 1999, 37, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Walker, A.S.; Peto, T.E.; Crook, D.W.; Wilson, D.J. Within-host evolution of bacterial pathogens. Nat. Rev. Microbiol. 2016, 14, 150–162. [Google Scholar] [CrossRef]
- Okoro, C.K.; Kingsley, R.A.; Quail, M.A.; Kankwatira, A.M.; Feasey, N.A.; Parkhill, J.; Dougan, G.; Gordon, M.A. High-resolution single nucleotide polymorphism analysis distinguishes recrudescence and reinfection in recurrent invasive nontyphoidal Salmonella typhimurium disease. Clin. Infect. Dis. 2012, 54, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Mac Aogain, M.; Moloney, G.; Kilkenny, S.; Kelleher, M.; Kelleghan, M.; Boyle, B.; Rogers, T.R. Whole-genome sequencing improves discrimination of relapse from reinfection and identifies transmission events among patients with recurrent Clostridium difficile infections. J. Hosp. Infect. 2015, 90, 108–116. [Google Scholar] [CrossRef]
- Miragaia, M.; Thomas, I.C.; Couto, I.; Enright, M.C.; de Lencastre, H. Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J. Bacteriol. 2007, 189, 2540–2552. [Google Scholar] [CrossRef]
- McAdam, P.R.; Holmes, A.; Templeton, K.E.; Fitzgerald, J.R. Adaptive evolution of Staphylococcus aureus during chronic endobronchial infection of a cystic fibrosis patient. PLoS ONE 2011, 6, e24301. [Google Scholar] [CrossRef]
- Hirschhausen, N.; Block, D.; Bianconi, I.; Bragonzi, A.; Birtel, J.; Lee, J.C.; Dubbers, A.; Kuster, P.; Kahl, J.; Peters, G.; et al. Extended Staphylococcus aureus persistence in cystic fibrosis is associated with bacterial adaptation. Int. J. Med. Microbiol. 2013, 303, 685–692. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Deming, C.; Conlan, S.; Program, N.C.S.; Barnabas, B.; Blakesley, R.; Bouffard, G.; Brooks, S.; Coleman, H.; et al. Biogeography and individuality shape function in the human skin metagenome. Nature 2014, 514, 59. [Google Scholar] [CrossRef]
- Rijnders, B.J.; Van Wijngaerden, E.; Van Eldere, J.; Peetermans, W.E. Polyclonal Staphylococcus epidermidis intravascular catheter-related infections. Clin. Microbiol. Infect. 2001, 7, 388–391. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sheppard, S.K.; Jolley, K.A.; Maiden, M.C. A Gene-By-Gene Approach to Bacterial Population Genomics: Whole Genome MLST of Campylobacter. Genes 2012, 3, 261–277. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate Source | No. Patients | No. Isolates | No. Patients with Paired Isolates | No. Patients with 3 or More Isolates | Time between Matched Paired Isolates (in Days) | |||
|---|---|---|---|---|---|---|---|---|
| (n = 55) | (n = 115) | (No. Isolates) | (No. Isolates) | <30 | 31–90 | 91–180 | >181 | |
| Prosthetic joint infection (PJI) | 50 | 103 | 48 (96) | 2 (7) | 13 | 17 | 20 | 3 |
| Fracture-related infection (FRI) a | 5 | 12 | 4 (8) | 1 (4) | 2 | 0 | 1 | 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harris, L.G.; Bodger, O.; Post, V.; Mack, D.; Morgenstern, M.; Rohde, H.; Moriarty, T.F.; Wilkinson, T.S. Temporal Changes in Patient-Matched Staphylococcus epidermidis Isolates from Infections: towards Defining a ‘True’ Persistent Infection. Microorganisms 2020, 8, 1508. https://doi.org/10.3390/microorganisms8101508
Harris LG, Bodger O, Post V, Mack D, Morgenstern M, Rohde H, Moriarty TF, Wilkinson TS. Temporal Changes in Patient-Matched Staphylococcus epidermidis Isolates from Infections: towards Defining a ‘True’ Persistent Infection. Microorganisms. 2020; 8(10):1508. https://doi.org/10.3390/microorganisms8101508
Chicago/Turabian StyleHarris, Llinos G., Owen Bodger, Virginia Post, Dietrich Mack, Mario Morgenstern, Holger Rohde, T. Fintan Moriarty, and Thomas S. Wilkinson. 2020. "Temporal Changes in Patient-Matched Staphylococcus epidermidis Isolates from Infections: towards Defining a ‘True’ Persistent Infection" Microorganisms 8, no. 10: 1508. https://doi.org/10.3390/microorganisms8101508
APA StyleHarris, L. G., Bodger, O., Post, V., Mack, D., Morgenstern, M., Rohde, H., Moriarty, T. F., & Wilkinson, T. S. (2020). Temporal Changes in Patient-Matched Staphylococcus epidermidis Isolates from Infections: towards Defining a ‘True’ Persistent Infection. Microorganisms, 8(10), 1508. https://doi.org/10.3390/microorganisms8101508