Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines

 ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Sample Collection
2.2. Factors that may Influence Bacterial Diversity in Washing Machines
2.3. DNA-Extraction
2.4. PCR and Clean Up
2.5. Pyrosequencing
2.6. Bioinformatic and Statistical Analyses
3. Results
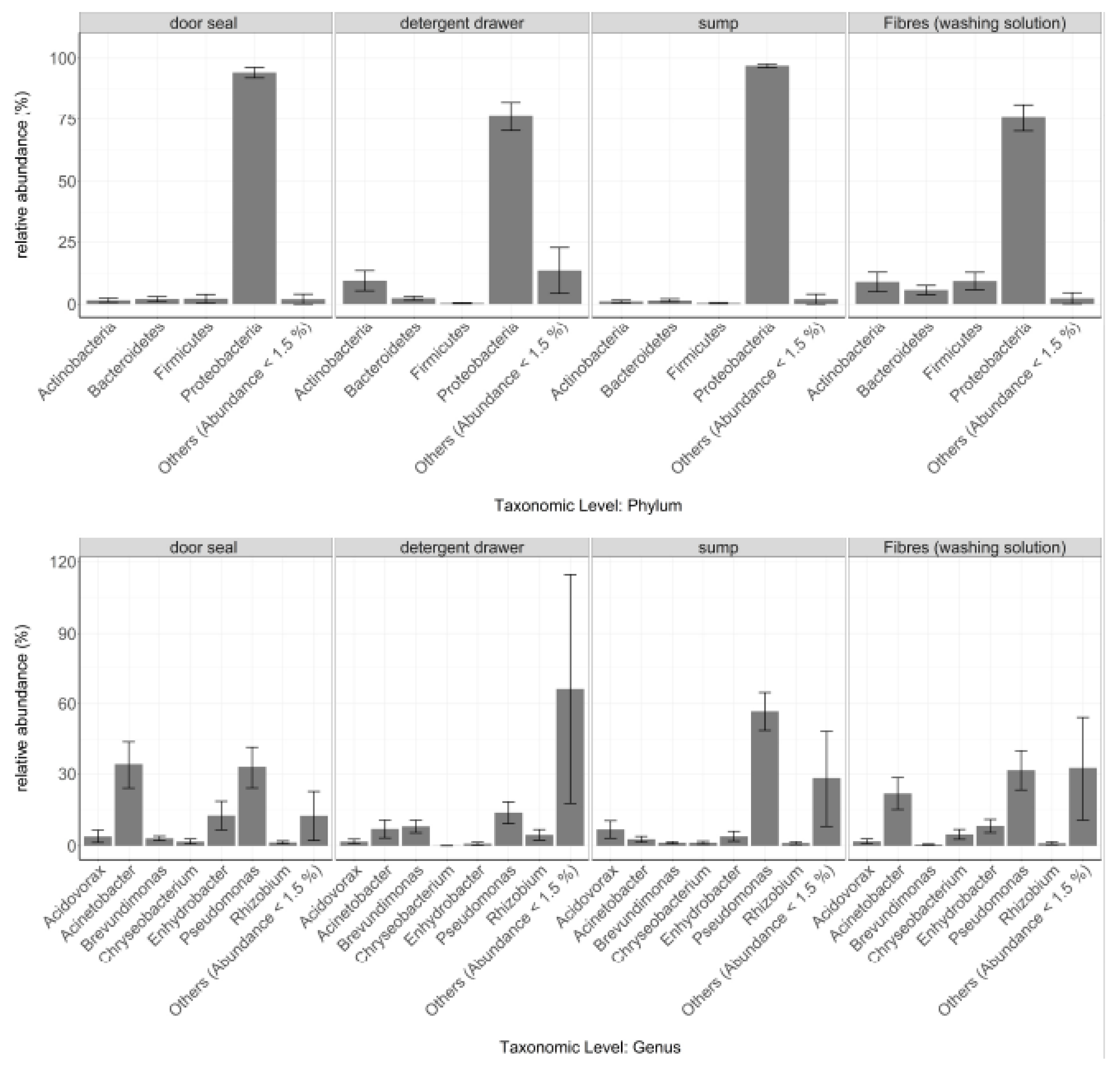
3.1. General Bacterial Community Composition
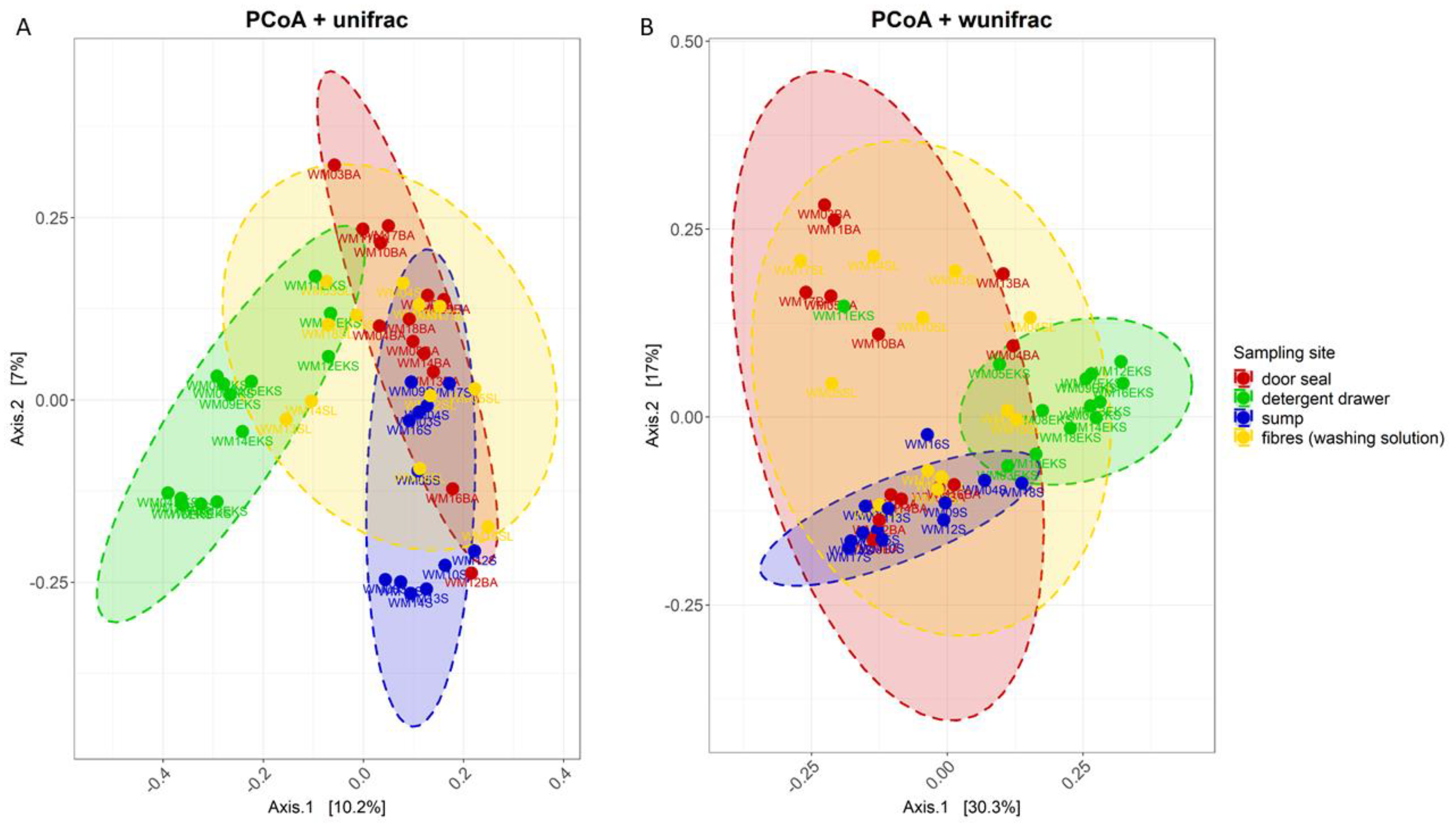
3.2. Site-Dependent Bacterial Community Composition
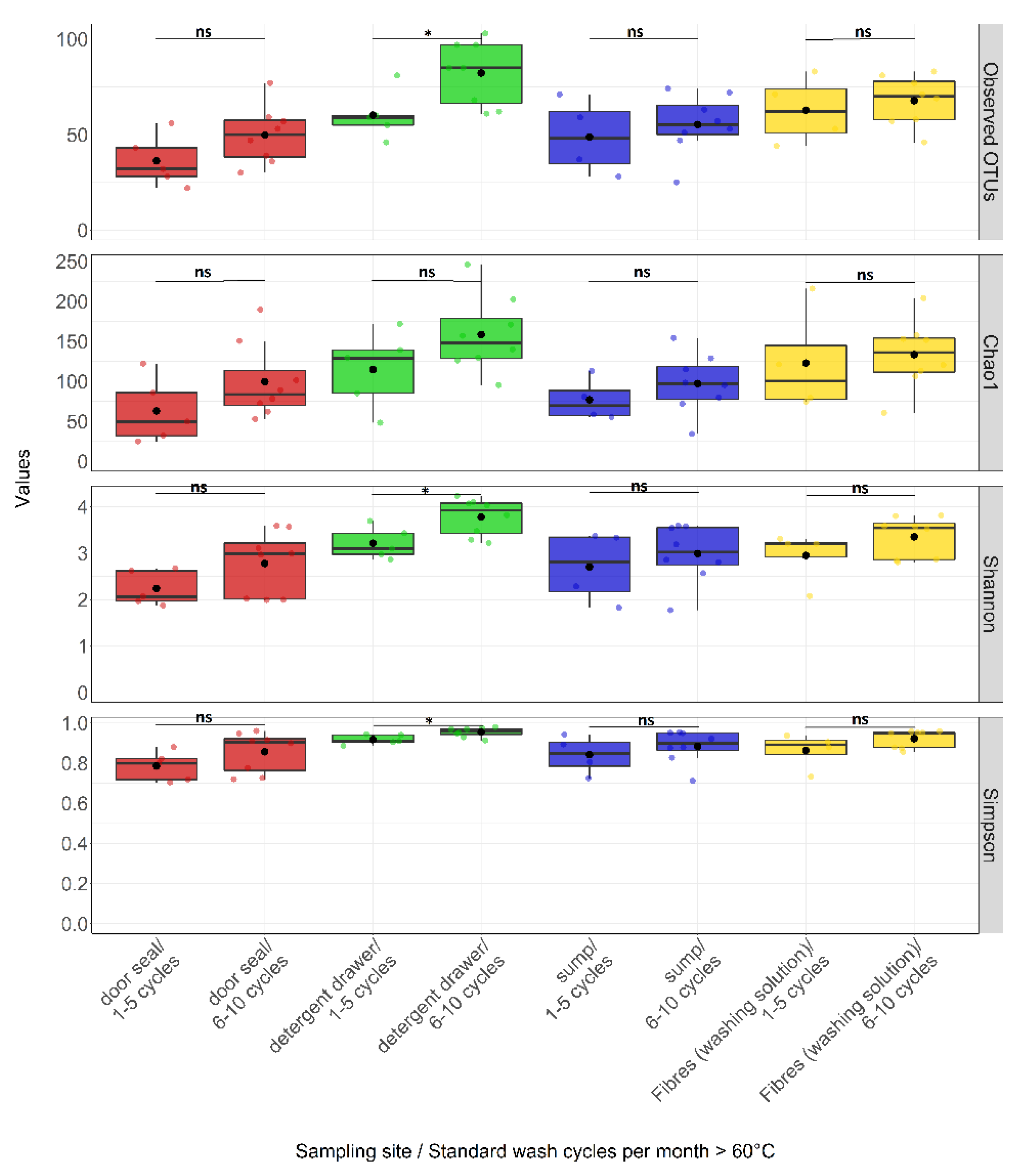
3.3. Effect of Environmental Factors on Community Composition
4. Discussion
4.1. The structure of the Bacterial Community Differs between Various Sampling Sites
4.2. Factors Influencing Bacterial Community Composition
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Relative Abundance (%) | SD | Positive Samples | OTU-ID | SILVA Genus | Top-hit taxon EzBioCloud | EzBioCloud Accession Number | Similarity (%) | Completeness (%) | RG (TRBA) |
|---|---|---|---|---|---|---|---|---|---|---|
| Door seal | 12.1 | 14.8 | 11 | denovo7218 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.3 | 1 |
| 10.1 | 13.3 | 11 | denovo5159 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.2 | 1 | |
| 7.2 | 14.6 | 8 | denovo7377 | Acinetobacter | JF232448_s | JF232448 | 98.97 | 33.3 | - | |
| 6.0 | 11.2 | 9 | denovo6937 | Enhydrobacter | Moraxella osloensis | APQL01000005 | 100.00 | 33.6 | 2 | |
| 4.1 | 13.5 | 2 | denovo3157 | Acinetobacter | Acinetobacter beijerinckii | APQL01000005 | 99.38 | 33.3 | 2 | |
| 3.8 | 6.7 | 7 | denovo301 | Enhydrobacter | Moraxella osloensis | CP014234 | 100.00 | 33.1 | 2 | |
| 3.6 | 8.3 | 7 | denovo6136 | Acinetobacter | Acinetobacter parvus | AIEB01000124 | 100.00 | 33.4 | 2 | |
| 3.4 | 8.9 | 5 | denovo4753 | Acinetobacter | Acinetobacter parvus | AIEB01000124 | 100.00 | 33.3 | 2 | |
| 3.1 | 7.3 | 5 | denovo3836 | Acidovorax | Acidovorax radicis | AFBG01000030 | 99.59 | 33.2 | 1 | |
| 2.7 | 5.9 | 7 | denovo3896 | - | Rhizobium rosettiformans | EU781656 | 98.91 | 32.6 | 1 | |
| Detergent drawer | 2.5 | 6.6 | 4 | denovo7377 | Acinetobacter | JF232448_s | JF232448 | 98.97 | 33.3 | - |
| 2.3 | 4.3 | 10 | denovo15 | Brevundimonas | Brevundimonas vesicularis | BCWM01000033 | 98.48 | 33.5 | 2 | |
| 2.1 | 4.5 | 4 | denovo8752 | - | Pseudoxanthomonas mexicana | AF273082 | 100.00 | 33.5 | 1 | |
| 1.9 | 7.0 | 1 | denovo6661 | uncultured | HQ856368_s | HQ856368 | 100.00 | 32.4 | - | |
| 1.9 | 5.3 | 3 | denovo1071 | Pseudomonas | Pseudomonas aeruginosa | BAMA01000316 | 99.00 | 34.2 | 2 | |
| 1.9 | 5.7 | 6 | denovo5179 | - | Pseudoxanthomonas mexicana | AF273082 | 99.20 | 34.6 | 1 | |
| 1.7 | 3.9 | 5 | denovo7389 | Rhizobium | Rhizobium radiobacter | AJ389904 | 98.47 | 32.8 | 1 | |
| 1.5 | 3.3 | 7 | denovo2452 | Brevundimonas | Brevundimonas vesicularis | BCWM01000033 | 100.00 | 31.4 | 2 | |
| 1.4 | 3.1 | 4 | denovo8373 | Aureimonas | Aureimonas altamirensis | BBWQ01000019 | 98.26 | 33.0 | 1 | |
| 1.4 | 4.9 | 1 | denovo5170 | Pseudomonas | Pseudomonas avellanae | AKBS01001374 | 100.00 | 33.4 | 1 | |
| Sump | 20.1 | 14.0 | 12 | denovo7218 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.3 | 1 |
| 17.0 | 11.8 | 12 | denovo5159 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.2 | 1 | |
| 4.7 | 9.0 | 5 | denovo3836 | Acidovorax | Acidovorax radicis | AFBG01000030 | 99.59 | 33.2 | 1 | |
| 2.6 | 5.0 | 5 | denovo2699 | Citrobacter | Citrobacter freundii | AJ233408 | 99.80 | 33.5 | 2 | |
| 2.3 | 3.0 | 8 | denovo4149 | - | Diaphorobacter nitroreducens | AB064317 | 99.59 | 33.3 | 1 | |
| 1.5 | 2.4 | 5 | denovo7180 | - | Diaphorobacter nitroreducens | AB064317 | 97.96 | 34.2 | 1 | |
| 1.5 | 2.9 | 6 | denovo6937 | Enhydrobacter | Moraxella osloensis | CP014234 | 100.00 | 33.6 | 2 | |
| 1.4 | 4.1 | 5 | denovo213 | Ochrobactrum | Ochrobactrum anthropi | CP000758 | 100.00 | 30.7 | 2 | |
| 1.2 | 2.6 | 5 | denovo301 | Enhydrobacter | Moraxella osloensis | CP014234 | 100.00 | 33.1 | 2 | |
| 1.2 | 2.0 | 4 | denovo6665 | - | Kosakonia sacchari | CP007215 | 99.80 | 33.4 | - | |
| Fibres (Washing solution) | 10.7 | 12.7 | 9 | denovo7218 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.3 | 1 |
| 10.3 | 12.4 | 10 | denovo5159 | Pseudomonas | Pseudomonas oleovorans subsp. oleovorans | NIUB01000072 | 100.00 | 33.2 | 1 | |
| 5.4 | 9.3 | 8 | denovo7377 | Acinetobacter | JF232448_s | JF232448 | 98.97 | 33.3 | - | |
| 3.0 | 3.9 | 9 | denovo301 | Enhydrobacter | Moraxella osloensis | CP014234 | 100.00 | 33.1 | 2 | |
| 2.9 | 4.9 | 5 | denovo7076 | Micrococcus | Micrococcus aloeverae | KF524364 | 100.00 | 32.3 | - | |
| 2.4 | 3.4 | 9 | denovo2637 | Acinetobacter | Acinetobacter johnsonii | APON01000005 | 98.97 | 33.4 | 2 | |
| 2.4 | 2.6 | 8 | denovo6937 | Enhydrobacter | Moraxella osloensis | CP014234 | 100.00 | 33.6 | 2 | |
| 2.1 | 3.7 | 6 | denovo1609 | Chryseobacterium | Chryseobacterium hominis | jgi.1096633 | 99.00 | 34.7 | 2 | |
| 2.0 | 6.9 | 1 | denovo5400 | Acinetobacter | Acinetobacter junii | APPX01000010 | 98.96 | 33.0 | 2 | |
| 1.8 | 2.7 | 7 | denovo2396 | Chryseobacterium | Chryseobacterium hominis | jgi.1096633 | 99.79 | 33.4 | 2 |
References
- Statistisches Jahrbuch Deutschland, 1st ed.; Statistisches Bundesamt: Wiesbaden, Germany, 2017.
- Bao, W.; Gong, R.H.; Ding, X.; Xue, Y.; Li, P.; Fan, W. Optimizing a laundering program for textiles in a front-loading washing machine and saving energy. J. Clean Prod. 2017, 148, 415–421. [Google Scholar] [CrossRef]
- Bloomfield, S.F.; Exner, M.; Signorelli, C.; Scott, E.A. Effectiveness of Laundering Processes Used in Domestic (Home) Settings; IFH: New York, NY, USA, 2013; pp. 1–62. Available online: https://www.ifh-homehygiene.org/system/files_force/publications/Effectiveness_of_laundering_IFHreport_21102013.pdf (accessed on 20 December 2019).
- Denawaka, C.J.; Fowlis, I.A.; Dean, J.R. Source, impact and removal of malodour from soiled clothing. J. Chromatogr. A 2016, 1438, 216–225. [Google Scholar] [CrossRef]
- Ossowski, B.; Duchmann, U. Der Einfluß des haushaltsüblichen Waschprozesses auf mykotisch kontaminierte Textilien. Der Hautarzt 1997, 48, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Bockmühl, D.P. Laundry hygiene-how to get more than clean. J. Appl. Microbiol. 2017, 122, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Egert, M. The BE-Microbiome-Communities with Relevance for Laundry and Home Care. SOFW J. 2017, 143, 44–48. [Google Scholar]
- Munk, S.; Johansen, C.; Stahnke, L.H.; Adler-Nissen, J. Microbial survival and odor in laundry. J. Surfact. Deterg. 2001, 4, 385–394. [Google Scholar] [CrossRef]
- Callewaert, C.; van Nevel, S.; Kerckhof, F.M.; Granitsiotis, M.; Boon, N. Bacterial Exchange in Household Washing Machines. Front. Microbiol. 2015, 6, 1381. [Google Scholar] [CrossRef]
- Bloomfield, S.; Exner, M.; Flemming, H.C.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Krämer, I.; Merkens, W.; Oltmanns, P.; et al. Lesser-known or hidden reservoirs of infection and implications for adequate prevention strategies: Where to look and what to look for. GMS Hyg. Infect. Control 2015, 10. [Google Scholar]
- Gupta, R.S. The phylogeny of proteobacteria: Relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol. Rev. 2000, 24, 367–402. [Google Scholar] [CrossRef]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef]
- Chandki, R.; Banthia, P.; Banthia, R. Biofilms: A microbial home. J. Indian Soc. Periodontol. 2011, 15, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, M.; Guerrero, R. Living together in biofilms: The microbial cell factory and its biotechnological implications. Microb. Cell Fact. 2016, 15, 165. [Google Scholar] [CrossRef] [PubMed]
- Brandwein, M.; Steinberg, D.; Meshner, S. Microbial biofilms and the human skin microbiome. NPJ Biofilms Microbiomes 2016, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, J.; Raghupathi, P.K.; Houf, K.; Burmølle, M.; Sørensen, S.J.; Gunde-Cimerman, N. Synergistic Interactions in Microbial Biofilms Facilitate the Establishment of Opportunistic Pathogenic Fungi in Household Dishwashers. Front. Microbiol. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Ulukanli, Z.; Digrak, M. Alkaliphilic Micro-organisms and Habitats. Turk. J. Biol. 2002, 26, 181–191. [Google Scholar]
- Ren, H.; Wang, W.; Liu, Y.; Liu, S.; Lou, L.; Cheng, D.; He, X.; Zhou, X.; Qiu, S.; Fu, L.; et al. Pyrosequencing analysis of bacterial communities in biofilms from different pipe materials in a city drinking water distribution system of East China. Appl. Microbiol. Biotechnol. 2015, 99, 10713–10724. [Google Scholar] [CrossRef]
- Gibson, L.L.; Rose, J.B.; Haas, C.N. Use of quantitative microbial risk assessment for evaluation of the benefits of laundry sanitation. Am. J. Infect. Control. 1999, 27, S34–S39. [Google Scholar] [CrossRef]
- Altenbaher, B.; Šostar Turk, S.; Fijan, S. Ecological parameters and disinfection effect of low-temperature laundering in hospitals in Slovenia. J. Clean Prod. 2011, 19, 253–258. [Google Scholar] [CrossRef]
- Babič, M.N.; Zalar, P.; Ženko, B.; Schroers, H.J.; Džeroski, S.; Gunde-Cimerman, N. Candida and Fusarium species known as opportunistic human pathogens from customer-accessible parts of residential washing machines. Fungal Biol. 2015, 119, 95–113. [Google Scholar] [CrossRef]
- Honisch, M.; Stamminger, R.; Bockmühl, D.P. Impact of wash cycle time, temperature and detergent formulation on the hygiene effectiveness of domestic laundering. J. Appl. Microbiol. 2014, 117, 1787–1797. [Google Scholar] [CrossRef]
- Nix, I.D.; Frontzek, A.; Bockmühl, D.P. Characterization of Microbial Communities in Household Washing Machines. TSD 2015, 52, 432–440. [Google Scholar] [CrossRef]
- Stapleton, K.; Hill, K.; Day, K.; Perry, J.D.; Dean, J.R. The potential impact of washing machines on laundry malodour generation. Lett. Appl. Microbiol. 2013, 56, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Teufel, L.; Pipal, A.; Schuster, K.C.; Staudinger, T.; Redl, B. Material-dependent growth of human skin bacteria on textiles investigated using challenge tests and DNA genotyping. J. Appl. Microbiol. 2010, 108, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Wiksell, J.C.; Pickett, M.S.; Hartman, P.A. Survival of microorganisms in laundered polyester-cotton sheeting. Appl. Microbiol. 1973, 25, 431–435. [Google Scholar]
- Pilloni, G.; Granitsiotis, M.S.; Engel, M.; Lueders, T. Testing the limits of 454 pyrotag sequencing: Reproducibility, quantitative assessment and comparison to T-RFLP fingerprinting of aquifer microbes. PLoS ONE 2012, 7, e40467. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package R package version 2.5-4. 2018. Available online: https://CRAN.R-project.org/package=vegan (accessed on 20 December 2019).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H.; Sievert, C. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- baua-Bundesanstalt für Arbeitsschutz und Arbeitsmedizin. TRBA 466, 2015. Available online: https://www.baua.de/DE/Angebote/Rechtstexte-und-Technische-Regeln/Regelwerk/TRBA/TRBA-466.html (accessed on 9 May 2019).
- Chiller, K.; Selkin, B.A.; Murakawa, G.J. Skin microflora and bacterial infections of the skin. J. Investig. Dermatol. Symp. Proc. 2001, 6, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Seong, C.N.; Kang, J.W.; Lee, J.H.; Seo, S.Y.; Woo, J.J.; Park, C.; Bae, K.S.; Kim, M.S. Taxonomic hierarchy of the phylum Firmicutes and novel Firmicutes species originated from various environments in Korea. J. Microbiol. Biotechnol. 2018, 56, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.M.; Hills, J.; Driscoll, K.; Fergus, D.J.; Grunden, A.M.; Dunn, R.R. Microbial diversity of extreme habitats in human homes. PeerJ 2016, 4, e2376. [Google Scholar] [CrossRef]
- Rojas-Herrera, R.A.; Ramos-Castillo, A.S.; Estrada-Medina, H.; De los Santos-Briones, C.; de Keb-Llanes, M.A.; Barrientos-Medina, R.C.; Peña-Ramírez, Y.G.; O’Connor, A. Living with detergents: Pyrosequencing-based assessment of bacterial community structures in soils subjected for decades to contamination by detergents. Ann. Microbiol. 2015, 65, 1313–1322. [Google Scholar] [CrossRef]
- Becerra-Castro, C.; Macedo, G.; Silva, A.M.T.; Manaia, C.M.; Nunes, O.C. Proteobacteria become predominant during regrowth after water disinfection. Sci. Total Environ. 2016, 573, 313–323. [Google Scholar] [CrossRef]
- Vaz-Moreira, I.; Nunes, O.C.; Manaia, C.M. Ubiquitous and persistent Proteobacteria and other Gram-negative bacteria in drinking water. Sci. Total Environ. 2017, 586, 1141–1149. [Google Scholar] [CrossRef]
- Kwon, S.; Moon, E.; Kim, T.S.; Hong, S.; Park, H.D. Pyrosequencing Demonstrated Complex Microbial Communities in a Membrane Filtration System for a Drinking Water Treatment Plant. Microbes. Environ. 2011, 26, 149–155. [Google Scholar] [CrossRef]
- McLellan, S.L.; Fisher, J.C.; Newton, R.J. The microbiome of urban waters. Int. Microbiol. 2015, 18, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Al-Khoja, M.S.; Darrell, J.H. The skin as the source of Acinetobacter and Moraxella species occurring in blood cultures. J. Clin. Pathol. 1979, 32, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Ring, H.C.; Thorsen, J.; Saunte, D.M.; Lilje, B.; Bay, L.; Riis, P.T.; Larsen, N.; Andersen, L.O.; Nielsen, H.V.; Miller, I.M.; et al. The Follicular Skin Microbiome in Patients with Hidradenitis Suppurativa and Healthy Controls. JAMA Dermatol. 2017, 153, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, N.J.; Younis, M.H.; Alobaidi, A.S.; Shaath, N.M. An unusual osteomyelitis caused by Moraxella osloensis: A case report. Int. J. Surg. Case Rep. 2017, 41, 146–149. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.S.; Ruth, A.; Coffin, S.E. Infection due to Moraxella osloensis: Case report and review of the literature. Clin. Infect. Dis. 2000, 30, 179–181. [Google Scholar] [CrossRef]
- Wong, D.; Nielsen, T.B.; Bonomo, R.A.; Pantapalangkoor, P.; Luna, B.B. Clinical and Pathophysiological Overview of Acinetobacter Infections: A Century of Challenges. Clin. Microbiol. Rev. 2017, 30, 409–447. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yabuki, M.; Hasegawa, Y. Review of odorants in human axillary odour and laundry malodour: The importance of branched C7 chain analogues in malodours perceived by humans. Flavour Fragr. J. 2013, 28, 223–230. [Google Scholar] [CrossRef]
- Kubota, H.; Mitani, A.; Niwano, Y.; Takeuchi, K.; Tanaka, A.; Yamaguchi, N.; Kawamura, Y.; Hitomi, J. Moraxella species are primarily responsible for generating malodor in laundry. Appl. Environ. Microbiol. 2012, 78, 3317–3324. [Google Scholar] [CrossRef]
- Cardinale, M.; Kaiser, D.; Lueders, T.; Schnell, S.; Egert, M. Microbiome analysis and confocal microscopy of used kitchen sponges reveal massive colonization by Acinetobacter, Moraxella and Chryseobacterium species. Sci. Rep. 2017, 7, 5791. [Google Scholar] [CrossRef]



| Influencing Factor | Levels | n | Observed | Chao1 | Shannon | Simpson | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Min | Max | Mean | SD | Min | Max | Mean | SD | Min | Max | Mean | SD | Min | Max | |||
| Site | Door seal | 13 | 44.54 | 15.50 | 22.00 | 77.00 | 85.44 | 46.88 | 25.00 | 189.80 | 2.57 | 0.63 | 1.88 | 3.59 | 0.83 | 0.09 | 0.70 | 0.96 |
| Detergent drawer | 13 | 73.77 | 18.49 | 46.00 | 103.00 | 141.64 | 51.03 | 48.15 | 246.00 | 3.56 | 0.46 | 2.87 | 4.24 | 0.94 | 0.03 | 0.89 | 0.98 | |
| Sump | 12 | 53.08 | 16.47 | 25.00 | 74.00 | 90.37 | 33.98 | 34.17 | 154.09 | 2.89 | 0.66 | 1.77 | 3.59 | 0.87 | 0.09 | 0.71 | 0.95 | |
| Fibres (washing solution) | 12 | 66.08 | 14.08 | 44.00 | 83.00 | 129.83 | 48.74 | 60.62 | 216.00 | 3.22 | 0.50 | 2.08 | 3.81 | 0.90 | 0.07 | 0.73 | 0.96 | |
| Age | 0 - 10 years | 34 | 58.85 | 20.96 | 22.00 | 103.00 | 113.79 | 55.82 | 25.00 | 246.00 | 3.03 | 0.73 | 1.83 | 4.24 | 0.88 | 0.09 | 0.70 | 0.98 |
| 11 -20 years | 16 | 60.44 | 16.74 | 25.00 | 97.00 | 107.85 | 39.38 | 34.17 | 171.88 | 3.14 | 0.53 | 1.77 | 4.07 | 0.90 | 0.06 | 0.71 | 0.97 | |
| Smell | Yes | 10 | 62.44 | 20.70 | 28.00 | 97.00 | 123.02 | 54.57 | 32.00 | 216.00 | 3.11 | 0.72 | 1.83 | 4.24 | 0.89 | 0.08 | 0.72 | 0.98 |
| No | 40 | 58.80 | 17.31 | 32.00 | 97.00 | 105.52 | 40.28 | 49.50 | 156.91 | 3.13 | 0.58 | 1.97 | 4.07 | 0.90 | 0.08 | 0.70 | 0.97 | |
| Standard wash cycles per month ≥60 °C | 1 - 5 cycles | 18 | 51.56 | 18.10 | 22.00 | 83.00 | 93.69 | 50.01 | 25.00 | 216.00 | 2.77 | 0.61 | 1.83 | 3.69 | 0.85 | 0.08 | 0.70 | 0.94 |
| 6 -10 cycles | 32 | 63.75 | 19.21 | 25.00 | 103.00 | 122.13 | 49.07 | 34.17 | 246.00 | 3.23 | 0.65 | 1.77 | 4.24 | 0.90 | 0.08 | 0.71 | 0.98 | |
| Regular Cleaning | Yes | 20 | 62.95 | 20.69 | 25.00 | 97.00 | 118.60 | 49.36 | 32.00 | 216.00 | 3.14 | 0.70 | 1.77 | 4.24 | 0.89 | 0.08 | 0.71 | 0.98 |
| No | 30 | 56.97 | 18.72 | 22.00 | 103.00 | 107.41 | 52.09 | 25.00 | 246.00 | 3.01 | 0.65 | 1.97 | 4.10 | 0.88 | 0.08 | 0.70 | 0.97 | |
| Softener | Yes | 16 | 62.44 | 20.70 | 28.00 | 97.00 | 123.02 | 54.57 | 32.00 | 216.00 | 3.11 | 0.72 | 1.83 | 4.24 | 0.89 | 0.08 | 0.72 | 0.98 |
| No | 34 | 57.91 | 19.12 | 22.00 | 103.00 | 106.65 | 48.89 | 25.00 | 246.00 | 3.04 | 0.65 | 1.77 | 4.10 | 0.88 | 0.08 | 0.70 | 0.97 | |
| Detergent | Liquid | 18 | 57.78 | 13.61 | 32.00 | 85.00 | 109.06 | 37.93 | 49.50 | 171.88 | 3.03 | 0.54 | 1.97 | 4.03 | 0.89 | 0.08 | 0.70 | 0.97 |
| Powder | 32 | 60.25 | 22.36 | 22.00 | 103.00 | 113.48 | 57.30 | 25.00 | 246.00 | 3.08 | 0.74 | 1.77 | 4.24 | 0.88 | 0.09 | 0.71 | 0.98 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacksch, S.; Kaiser, D.; Weis, S.; Weide, M.; Ratering, S.; Schnell, S.; Egert, M. Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines. Microorganisms 2020, 8, 30. https://doi.org/10.3390/microorganisms8010030
Jacksch S, Kaiser D, Weis S, Weide M, Ratering S, Schnell S, Egert M. Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines. Microorganisms. 2020; 8(1):30. https://doi.org/10.3390/microorganisms8010030
Chicago/Turabian StyleJacksch, Susanne, Dominik Kaiser, Severin Weis, Mirko Weide, Stefan Ratering, Sylvia Schnell, and Markus Egert. 2020. "Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines" Microorganisms 8, no. 1: 30. https://doi.org/10.3390/microorganisms8010030
APA StyleJacksch, S., Kaiser, D., Weis, S., Weide, M., Ratering, S., Schnell, S., & Egert, M. (2020). Influence of Sampling Site and other Environmental Factors on the Bacterial Community Composition of Domestic Washing Machines. Microorganisms, 8(1), 30. https://doi.org/10.3390/microorganisms8010030