Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Procedures
2.1.1. Sample Collection
2.1.2. DNA Extraction and Amplicon Preparation
2.1.3. Sequencing
2.2. Bioinformatic Methods
2.2.1. Quality Preprocessing
2.2.2. Clustering and Sequence Identification
2.2.3. Sparsity Reduction and Taxonomy
2.2.4. In Silico Sets of GreenGenes and Silva Databases for V3, V4 and V3V4 Regions
2.2.5. Recruitment Analyses
2.2.6. Standardized Tables
2.2.7. Diversity Analyses
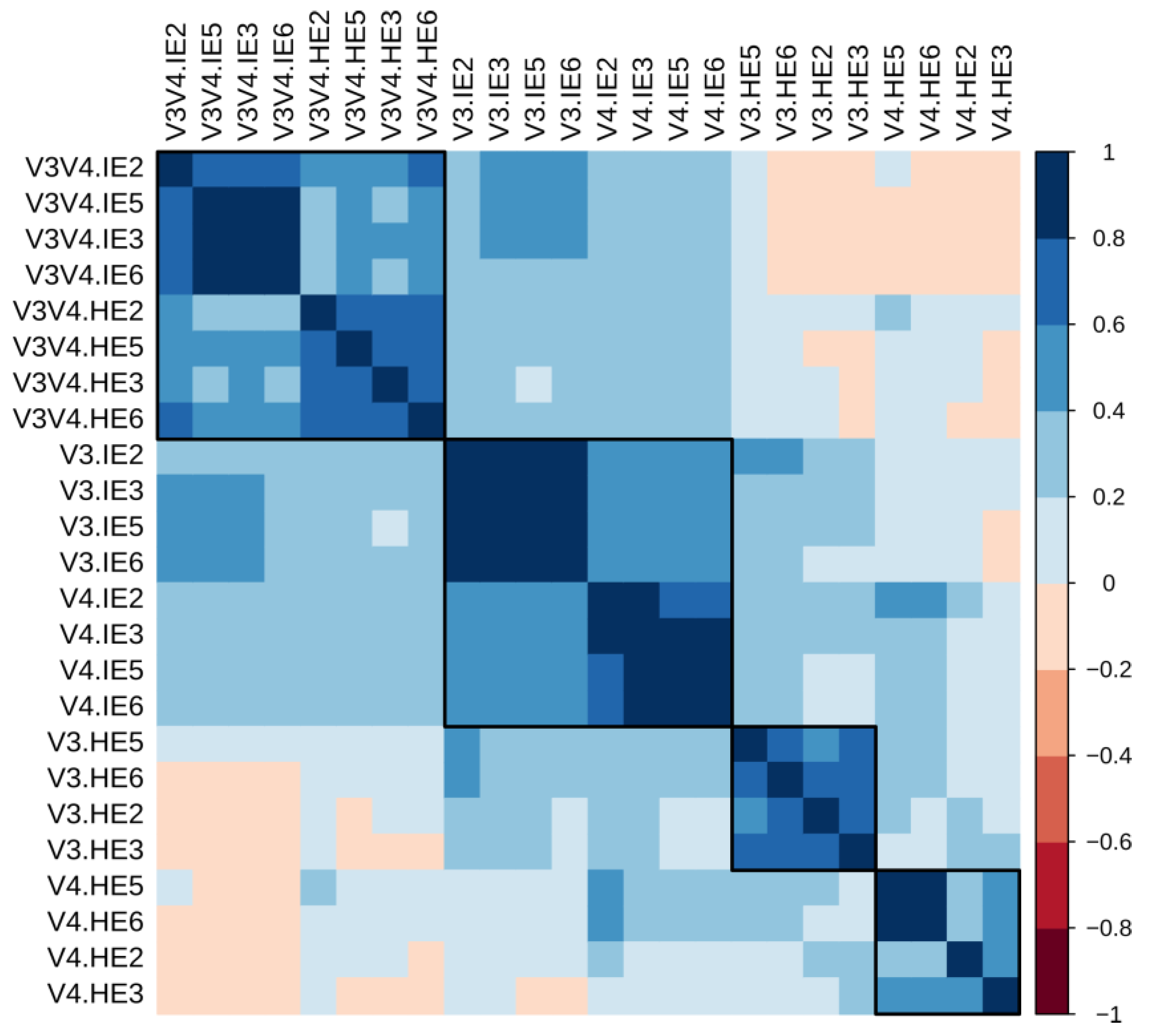
2.2.8. Sample Correlation Analysis
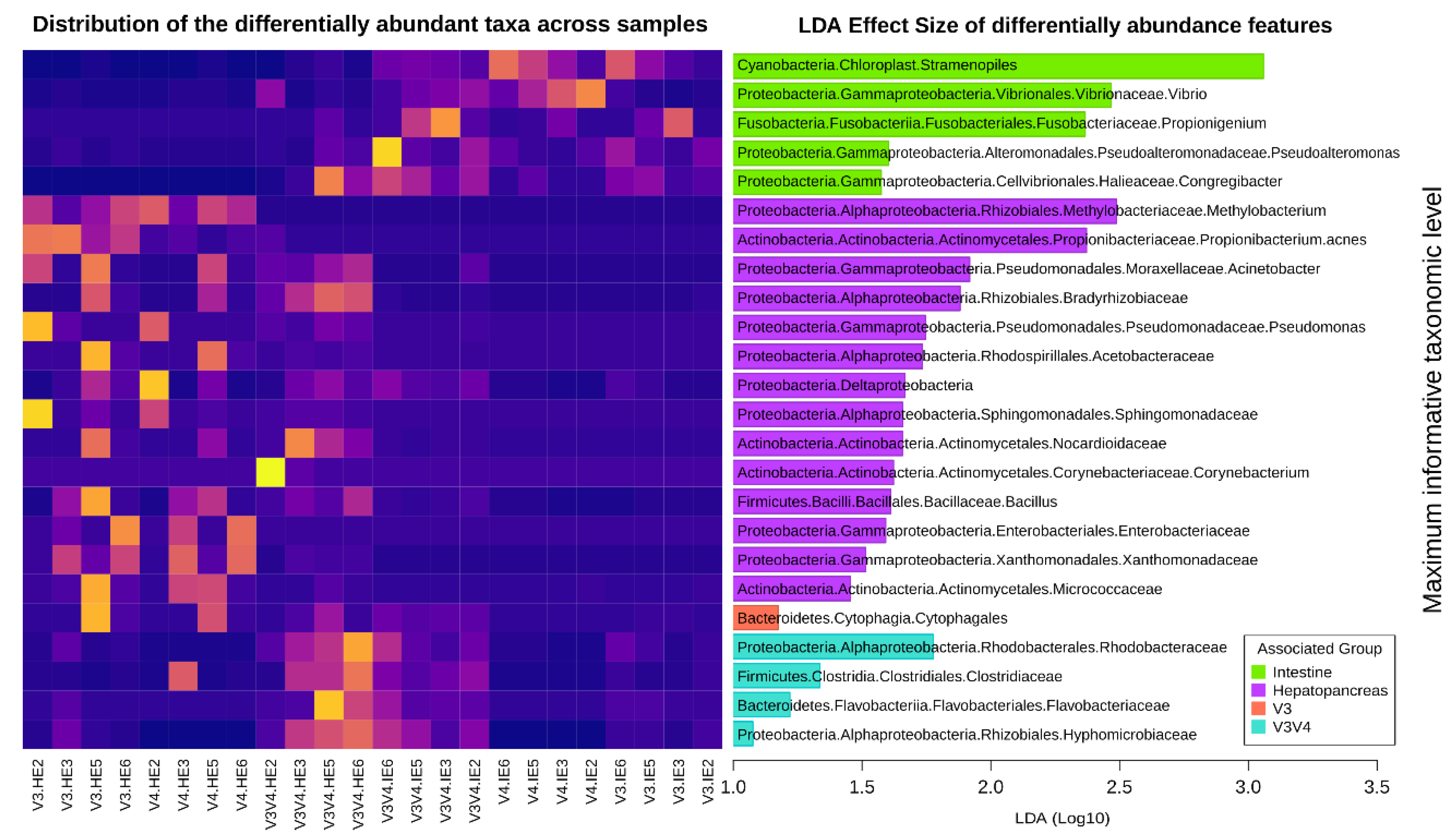
2.2.9. Differential Abundant Features
3. Results
3.1. Amplicon Sequencing of Biological Samples
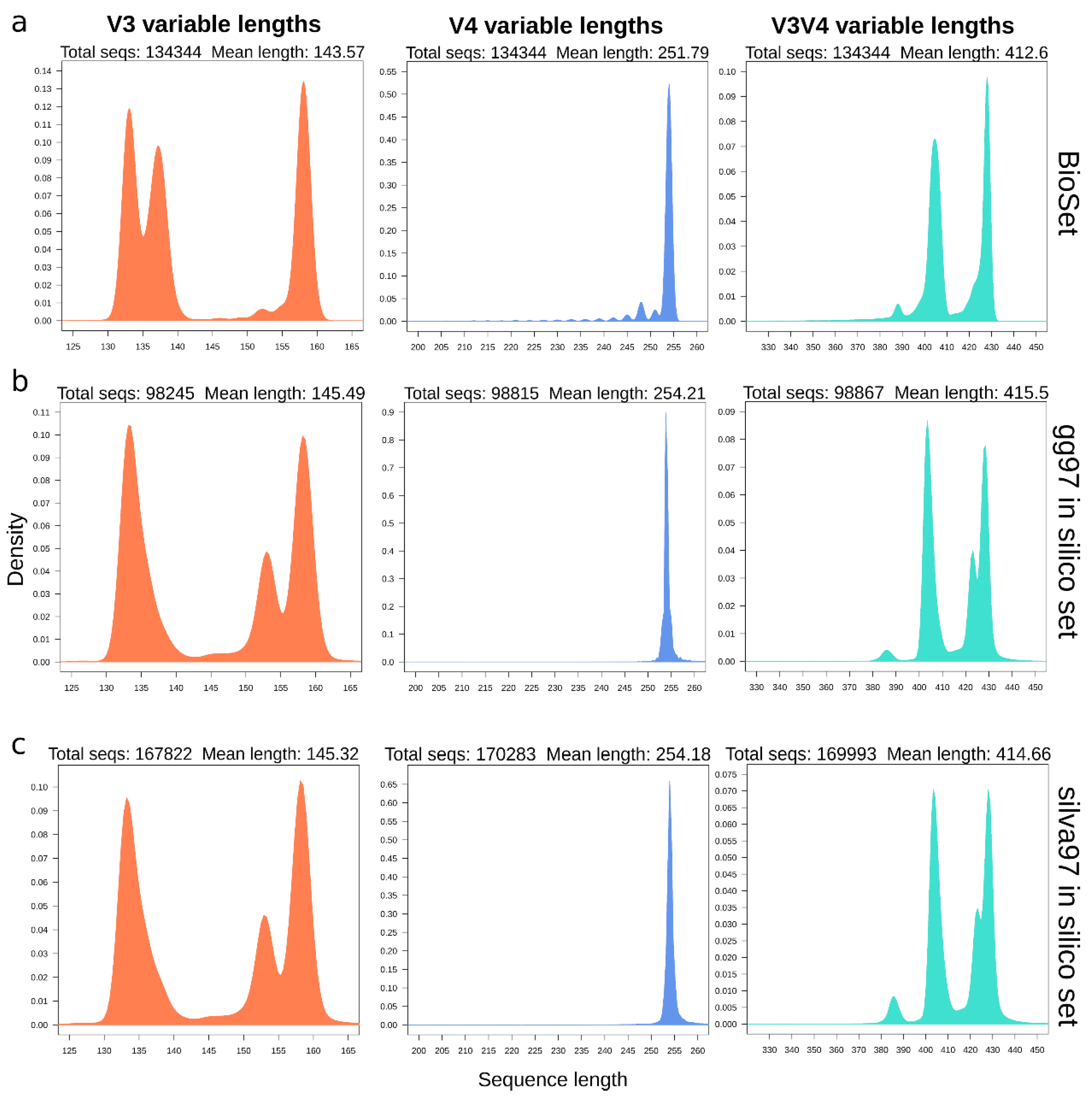
3.2. In Silico Amplicons Produce Length Distributions Similar to Experimental Ones
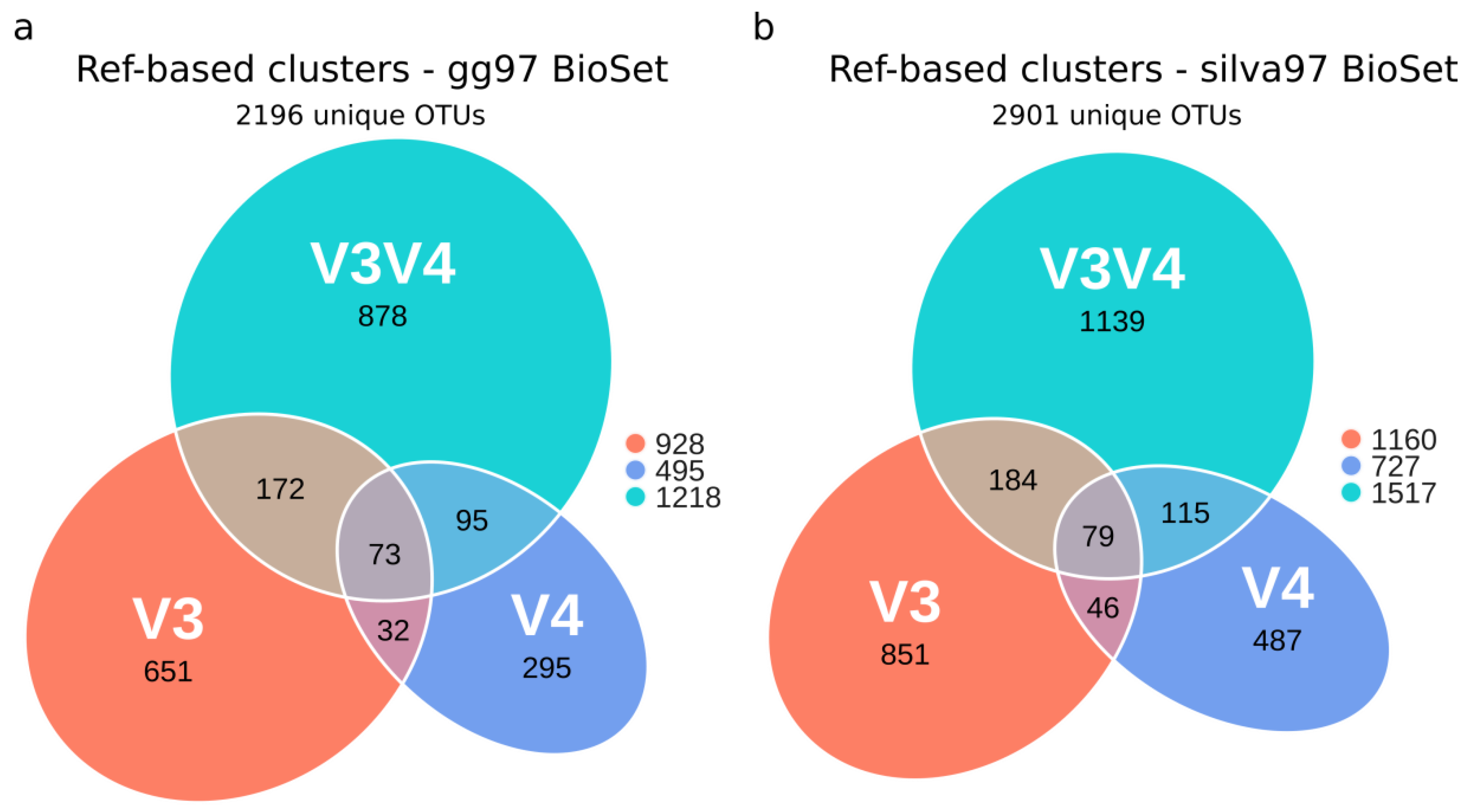
3.3. Evaluation of OTU’s Similarity between Regions
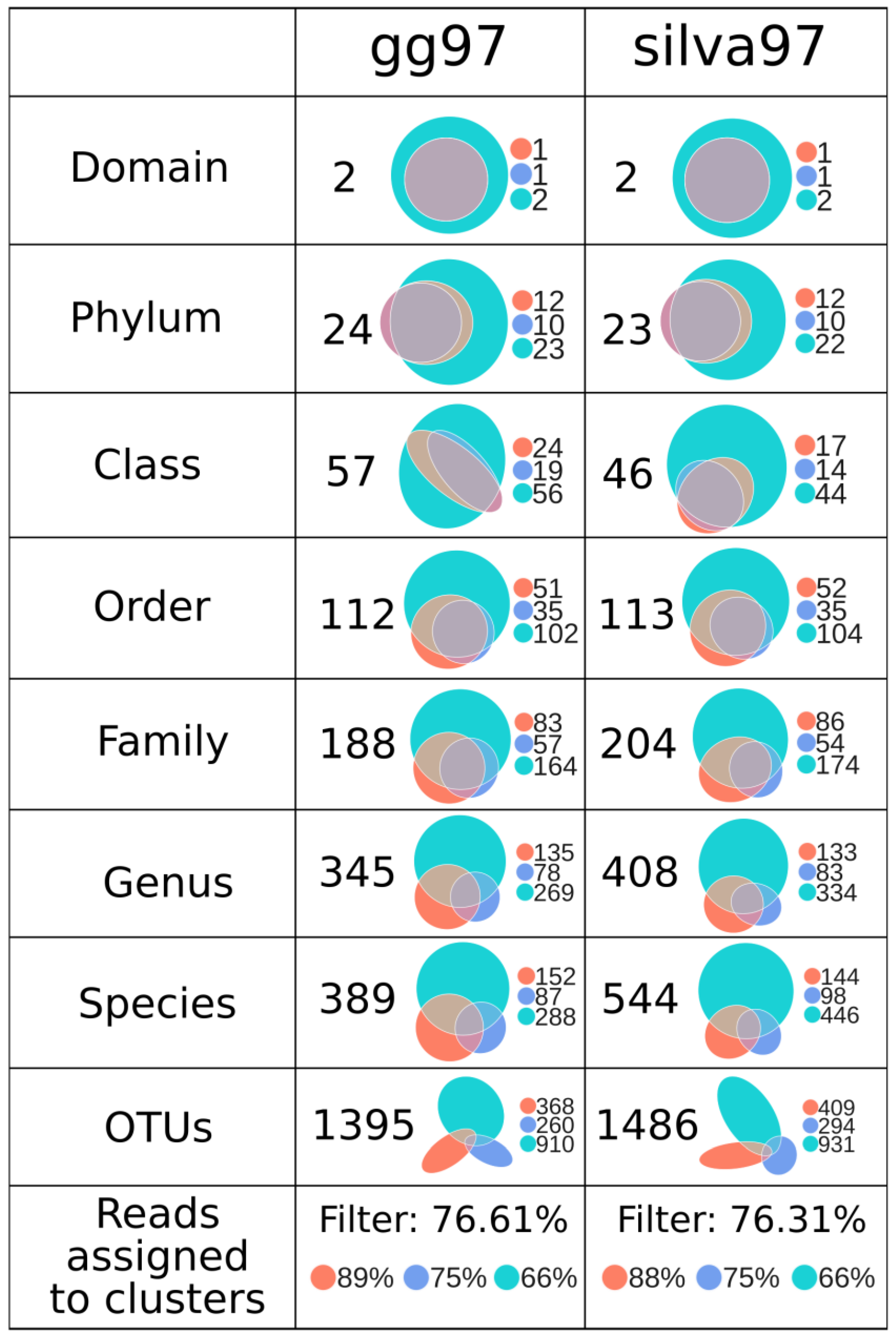
3.4. GreenGenes and Silva Capture a Similar Number of Low-Level Taxonomies
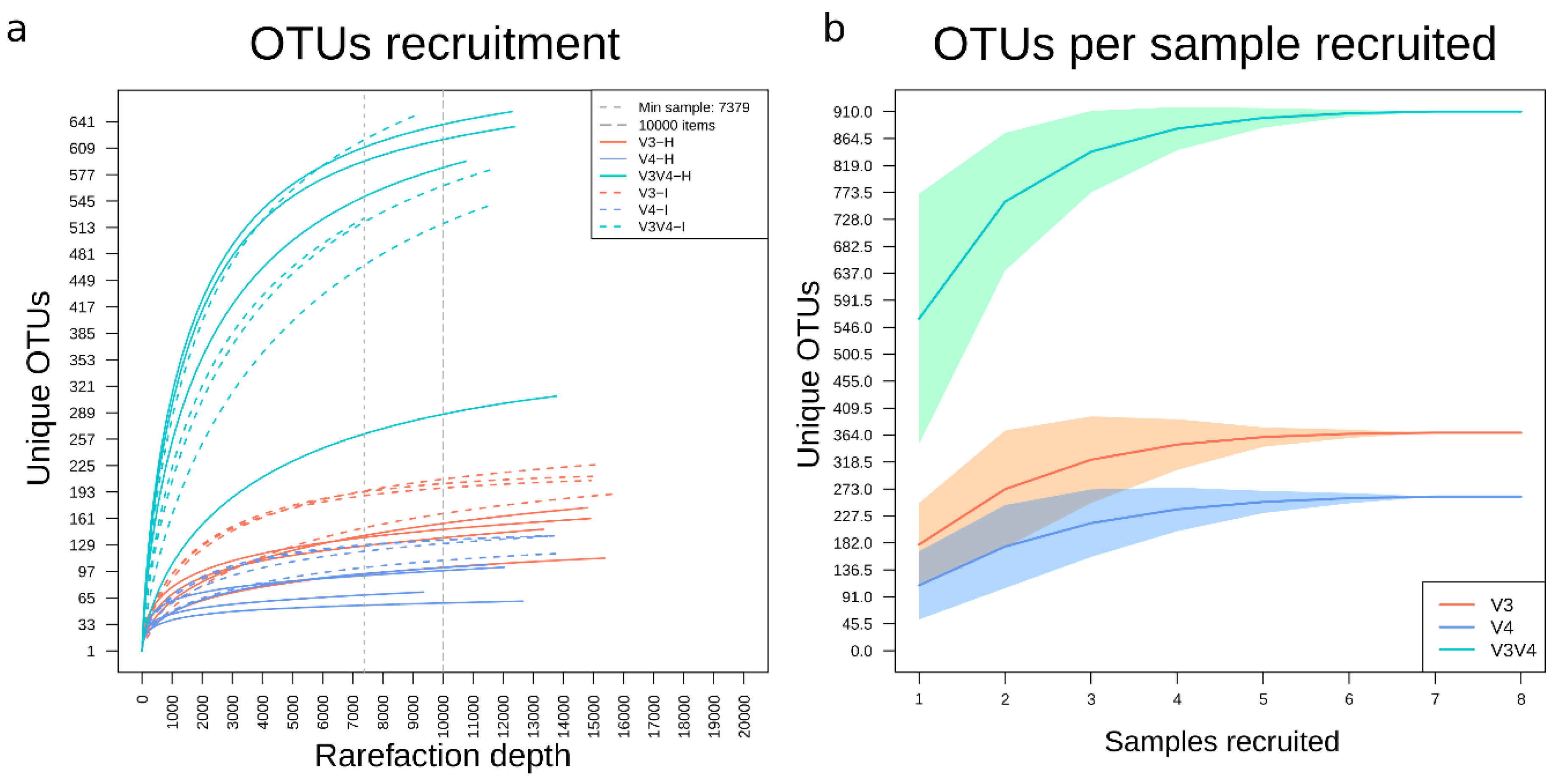
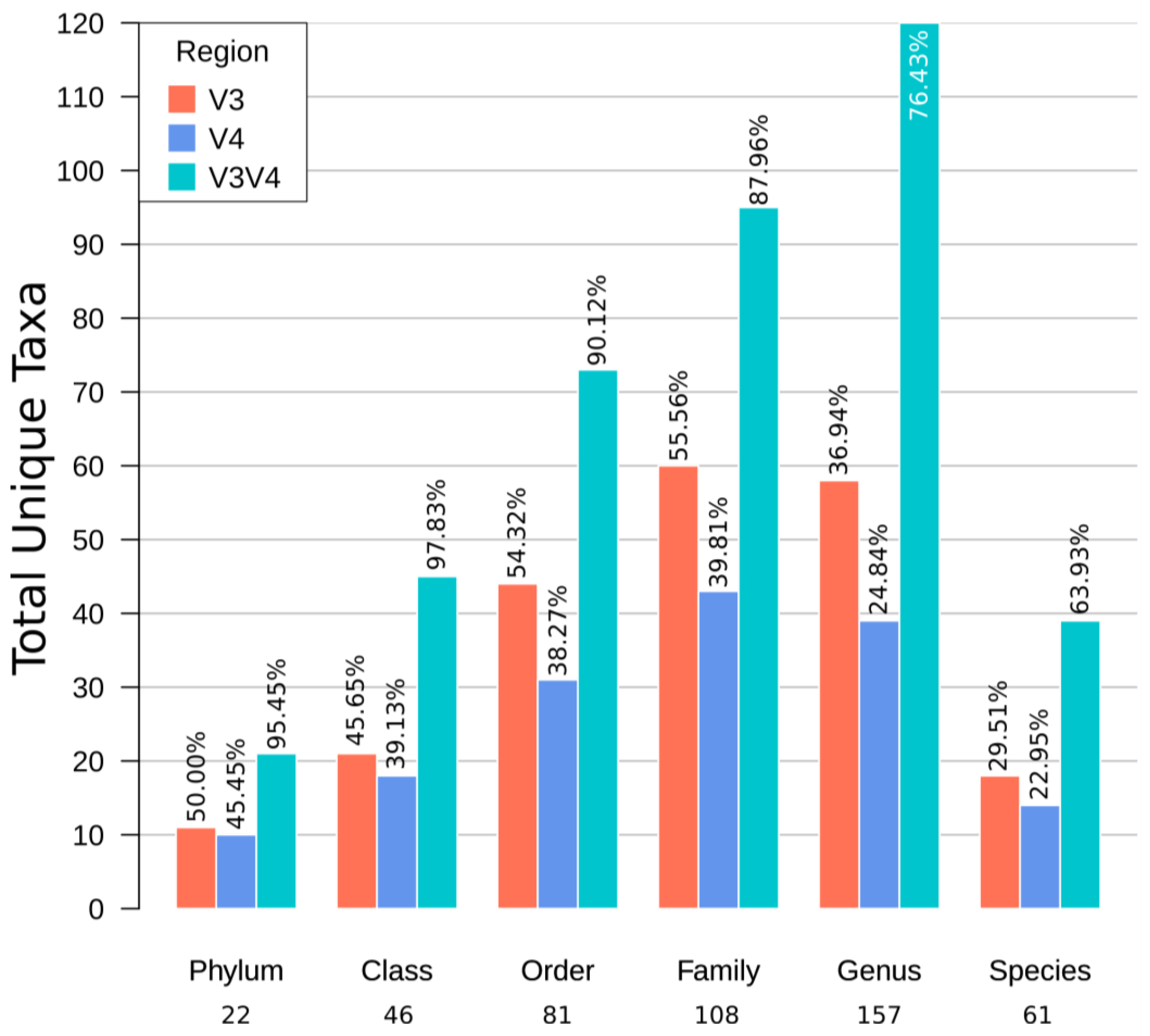
3.5. Recruitment Analyses Showed a Higher Number of Unique OTUs in V3V4 Followed by V3 and V4
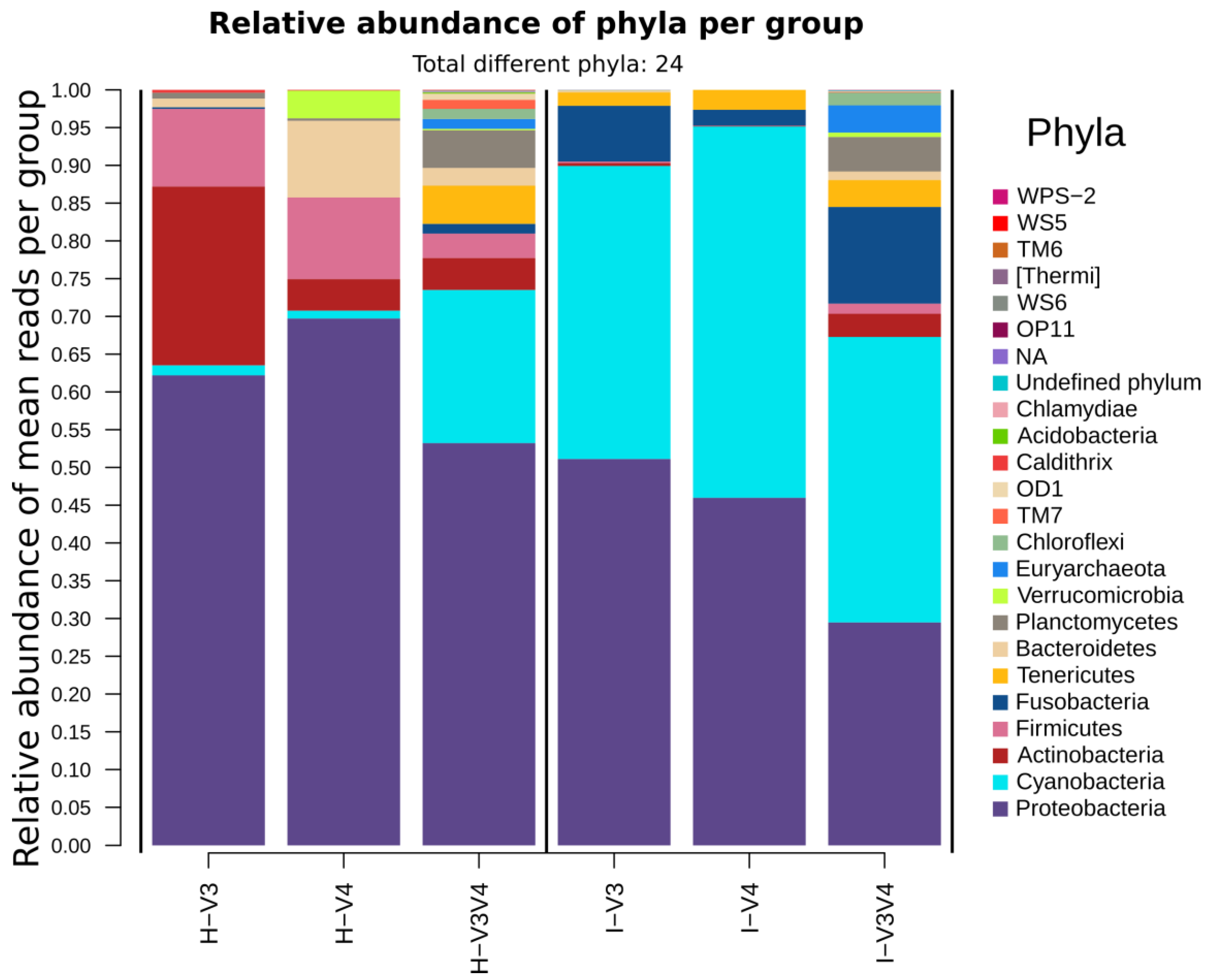
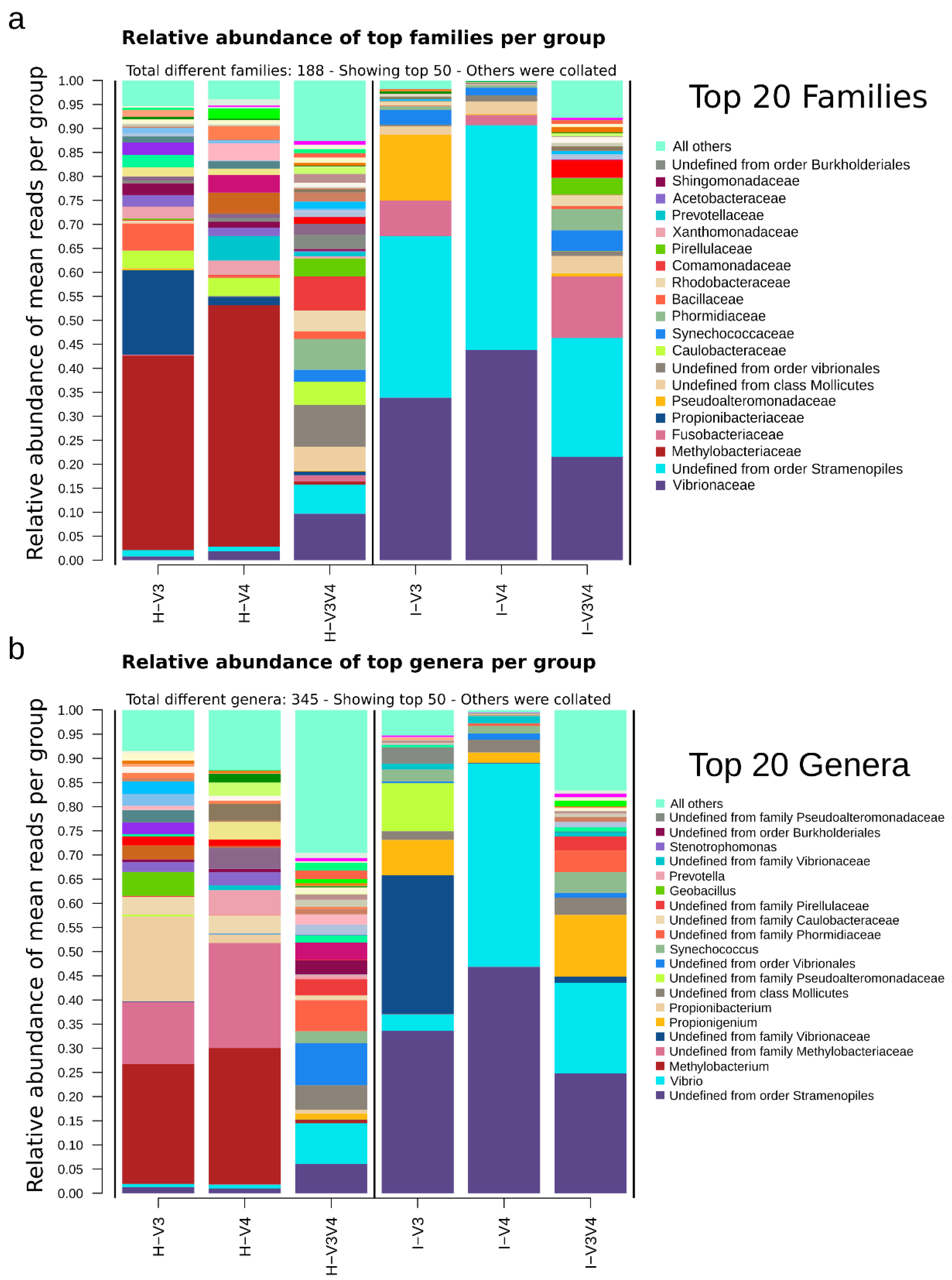
3.6. The Taxonomic Composition is Region and Organ Dependent
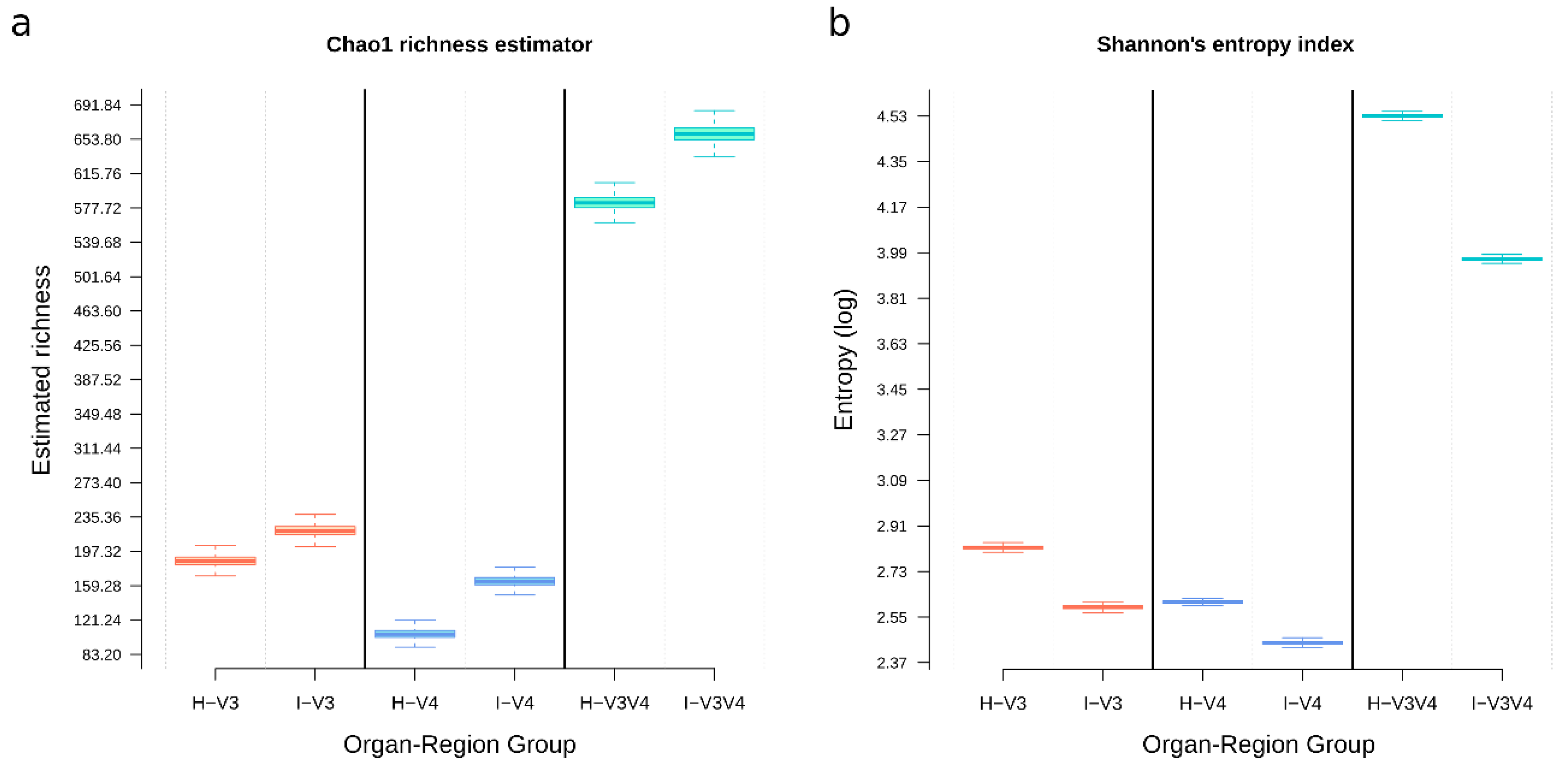
3.7. V3V4 Showed the More Considerable Richness and Diversity, Followed by V3 and V4
3.8. Beta Diversity Shows that Most Region-Organ Groups are Different in Terms of Composition and Abundance
3.9. Correlation Analyses Show that Sample Similarity is Heavily Influenced by Organ in the V3 and V4
3.10. Differential Abundance Analyses Detect More Features Specific to Organs than to Regions
4. Discussion
4.1. Experimental and Analysis Considerations
4.2. Microbiota Structure and Composition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO. FAO Yearbook. Fishery and Aquaculture Statistics 2017/FAO Annuaire. Statistiques des Pêches et de L’aquaculture 2017/FAO Anuario. Estadísticas de Pesca y Acuicultura 2017; FAO: Rome, Italy, 2019; ISBN 978-92-5-131669-6. [Google Scholar]
- FAO. Introductions and Movement of Penaeus Vannamei and Penaeus Stylirostris in Asia and the Pacific; FAO: Bangkok, Thailand, 2004. [Google Scholar]
- Perez-Enriquez, R.; Hernández-Martínez, F.; Cruz, P. Genetic diversity status of White shrimp Penaeus (Litopenaeus) vannamei broodstock in Mexico. Aquaculture 2009, 297, 44–50. [Google Scholar] [CrossRef]
- De Donato, M.; Manrique, R.; Ramirez, R.; Mayer, L.; Howell, C. Mass selection and inbreeding effects on a cultivated strain of Penaeus (Litopenaeus) vannamei in Venezuela. Aquaculture 2005, 247, 159–167. [Google Scholar] [CrossRef]
- Ghaffari, N.; Sanchez-Flores, A.; Doan, R.; Garcia-Orozco, K.D.; Chen, P.L.; Ochoa-Leyva, A.; Lopez-Zavala, A.A.; Carrasco, J.S.; Hong, C.; Brieba, L.G.; et al. Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci. Rep. 2014, 4, 7081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, J.; Sun, Y.; Li, S.; Gao, Y.; Yu, Y.; Liu, C.; Wang, Q.; Lv, X.; Zhang, X.; et al. Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat. Commun. 2019, 10, 356. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Granados, F.; Lopez-Zavala, A.A.; Gallardo-Becerra, L.; Mendoza-Vargas, A.; Sánchez, F.; Vichido, R.; Brieba, L.G.; Viana, M.T.; Sotelo-Mundo, R.R.; Ochoa-Leyva, A. Microbiome of Pacific Whiteleg shrimp reveals differential bacterial community composition between Wild, Aquacultured and AHPND/EMS outbreak conditions. Sci. Rep. 2017, 7, 11783. [Google Scholar] [CrossRef]
- Schock, T.B.; Duke, J.; Goodson, A.; Weldon, D.; Brunson, J.; Leffler, J.W.; Bearden, D.W. Evaluation of Pacific White Shrimp (Litopenaeus vannamei) Health during a Superintensive Aquaculture Growout Using NMR-Based Metabolomics. PLoS ONE 2013, 8, e59521. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, K.; Wu, J.; Qiuqian, L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Karoonuthaisiri, N. Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. J. Invertebr. Pathol. 2016, 133, 12–19. [Google Scholar] [CrossRef]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberón, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 2017, 19, 1490–1501. [Google Scholar] [CrossRef]
- Dai, W.F.; Zhang, J.J.; Qiu, Q.F.; Chen, J.; Yang, W.; Ni, S.; Xiong, J.B. Starvation stress affects the interplay among shrimp gut microbiota, digestion and immune activities. Fish Shellfish Immunol. 2018, 80, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Cornejo-Granados, F.; Gallardo-Becerra, L.; Leonardo-Reza, M.; Ochoa-Romo, J.P.; Ochoa-Leyva, A. A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 2018, 2018, e5382. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wang, Y.; Liu, Q.; Xiong, D.; Zhang, J. Transcriptomic and microbiota response on Litopenaeus vannamei intestine subjected to acute sulfide exposure. Fish Shellfish Immunol. 2019, 88, 335–343. [Google Scholar] [CrossRef]
- Chen, C.Y.; Chen, P.C.; Weng, F.C.H.; Shaw, G.T.W.; Wang, D. Habitat and indigenous gut microbes contribute to the plasticity of gut microbiome in oriental river prawn during rapid environmental change. PLoS ONE 2017, 12, e0181427. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, D.; Peatman, E.; Rhodes, M.A.; Liu, J.; Davis, D.A. Effects of various levels of dietary copper supplementation with copper sulfate and copper hydroxychloride on Pacific white shrimp Litopenaeus vannamei performance and microbial communities. Aquaculture 2017, 476, 94–105. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, Y.; Chen, K.; Yu, N.; Zhou, Z.; Chen, L.; Du, Z.; Li, E. Characterization of the intestinal microbiota in Pacific white shrimp, Litopenaeus vannamei, fed diets with different lipid sources. Aquaculture 2014, 434, 449–455. [Google Scholar] [CrossRef]
- Illumina, Inc. Illumina Sequencing Platforms. Available online: https://www.illumina.com/systems/sequencing-platforms.html (accessed on 16 October 2019).
- Mazón-Suástegui, J.M.; Salas-Leiva, J.S.; Medina-Marrero, R.; Medina-García, R.; García-Bernal, M. Effect of Streptomyces probiotics on the gut microbiota of Litopenaeus vannamei challenged with Vibrio parahaemolyticus. Microbiologyopen 2019. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Willis, C.; Desai, D.; Laroche, J. Influence of 16S rRNA variable region on perceived diversity of marine microbial communities of the Northern North Atlantic. FEMS Microbiol. Lett. 2019, 366, fnz152. [Google Scholar] [CrossRef]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16s rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed]
- Graspeuntner, S.; Loeper, N.; Künzel, S.; Baines, J.F.; Rupp, J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 2018, 8, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Farfante Perez, I.; Frederick Kensley, B. Penaeoid and Sergestoid Shrimps and Prawns of the World: Keys and Diagnoses for the Families and Genera, 1st ed.; Editions du Muséum: Paris, France, 1997; ISBN 2856535100 9782856535103. [Google Scholar]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Liu, B.; Yuan, J.; Yiu, S.M.; Li, Z.; Xie, Y.; Chen, Y.; Shi, Y.; Zhang, H.; Li, Y.; Lam, T.W.; et al. COPE: An accurate k-mer-based pair-end reads connection tool to facilitate genome assembly. Bioinformatics 2012, 28, 2870–2874. [Google Scholar] [CrossRef]
- Li, H. Seqtk Toolkit for Processing Sequences in FASTA/Q Formats. Available online: https://github.com/lh3/seqtk (accessed on 5 March 2019).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. silva: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Varoquaux, G.; Buitinck, L.; Louppe, G.; Grisel, O.; Pedregosa, F.; Mueller, A. Scikit-learn: Machine Learning Without Learning the Machinery. GetMobile 2015, 19, 29–33. [Google Scholar] [CrossRef]
- Larsson, J. Eulerr. Available online: https://github.com/jolars/eulerr (accessed on 17 October 2019).
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 March 2019).
- Jari Oksanen, F.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Peter, R.; Minchin, R.B.; O’Hara; Gavin, L.; et al. The Vegan Community Ecology Package. Available online: https://cran.r-project.org/web/packages/vegan/ (accessed on 1 March 2019).
- Janssen, S.; Mcdonald, D.; Gonzalez, A.; Navas-molina, J.A.; Jiang, L.; Xu, Z. Phylogenetic Placement of Exact Amplicon Sequences improves associations with clinical information. Msystems 2018, 3, e00021-18. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- NIH. Sequencing Costs. February 2019. Available online: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data (accessed on 16 October 2019).
- Van Helden, P. The cost of research in developing countries. EMBO Rep. 2012, 13, 395. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Schmidt, T.S.B.; Matias Rodrigues, J.F.; von Mering, C. Ecological Consistency of SSU rRNA-Based Operational Taxonomic Units at a Global Scale. PLoS Comput. Biol. 2014, 10, e1003594. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Hugenholtz, P.; Skarshewski, A.; Parks, D.H. Genome-based microbial taxonomy coming of age. Cold Spring Harb. Perspect. Biol. 2016, 8, a018085. [Google Scholar] [CrossRef]
- Soergel, D.A.W.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, X.; Guan, R.; Zhu, C.; Xu, C.; Zhu, B.; Zhang, H.; Xiong, Z.; Xue, Y.; Tu, J.; et al. Evaluation of different 16S rRNA gene V regions for exploring bacterial diversity in a eutrophic freshwater lake. Sci. Total Environ. 2018, 618, 1254–1267. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Balvočiute, M.; Huson, D.H. silva, RDP, Greengenes, NCBI and OTT—How do these taxonomies compare? BMC Genom. 2017, 18, 114. [Google Scholar] [CrossRef]
- Rőszer, T. The invertebrate midintestinal gland (“hepatopancreas”) is an evolutionary forerunner in the integration of immunity and metabolism. Cell Tissue Res. 2014, 358, 685–695. [Google Scholar] [CrossRef]
- Cheung, M.K.; Yip, H.Y.; Nong, W.; Law, P.T.W.; Chu, K.H.; Kwan, H.S.; Hui, J.H.L. Rapid Change of Microbiota Diversity in the Gut but Not the Hepatopancreas During Gonadal Development of the New Shrimp Model Neocaridina denticulata. Mar. Biotechnol. 2015, 17, 811–819. [Google Scholar] [CrossRef]
- McFadden, G.I. Primary and secondary endosymbiosis and the origin of plastids. J. Phycol. 2001, 37, 951–959. [Google Scholar] [CrossRef]
- Hanshew, A.S.; Mason, C.J.; Raffa, K.F.; Currie, C.R. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J. Microbiol. Methods 2013, 95, 149–155. [Google Scholar] [CrossRef]
- Moore, L.R.; Rocap, G.; Chisholm, S.W. Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature 1998, 393, 464–467. [Google Scholar] [CrossRef]
- Cardona, E.; Gueguen, Y.; Magré, K.; Lorgeoux, B.; Piquemal, D.; Pierrat, F.; Noguier, F.; Saulnier, D. Bacterial community characterization of water and intestine of the shrimp Litopenaeus stylirostris in a biofloc system. BMC Microbiol. 2016, 19, 157. [Google Scholar] [CrossRef]
- Wang, C.; Lin, G.; Yan, T.; Zheng, Z.; Chen, B.; Sun, F. The cellular community in the intestine of the shrimp Penaeus penicillatus and its culture environments. Fish Sci. 2014, 80, 1001–1007. [Google Scholar] [CrossRef]
- Dai, W.; Yu, W.; Zhang, J.; Zhu, J.; Tao, Z.; Xiong, J. The gut eukaryotic microbiota influences the growth performance among cohabitating shrimp. Appl. Microbiol. Biotechnol. 2017, 101, 6447–6457. [Google Scholar] [CrossRef] [PubMed]
- Hauer, T.; Komárek, J. CyanoDB.cz 2.0—On-Line Database of Cyanobacterial Genera. Available online: http://www.cyanodb.cz/ (accessed on 2 November 2019).
- Patt, T.E.; Cole, G.C.; Hanson, R.S. Methylobacterium, a New Genus of Facultatively Methylotrophic Bacteria. Int. J. Syst. Bacteriol. 1976, 26, 226–229. [Google Scholar] [CrossRef]
- Md Zoqratt, M.Z.H.; Eng, W.W.H.; Thai, B.T.; Austin, C.M.; Gan, H.M. Microbiome analysis of Pacific white shrimp gut and rearing water from Malaysia and Vietnam: Implications for aquaculture research and management. PeerJ 2018, 6, e5826. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.-R.; Rohde, M.; Bennasar, A. The Family Caulobacteraceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 179–205. ISBN 978-3-642-30197-1. [Google Scholar]
- Schink, B. The Genus Propionigenium. In The Prokaryotes; Springer: New York, NY, USA, 1992; pp. 3948–3951. [Google Scholar]
- Zheng, Y.; Yu, M.; Liu, J.; Qiao, Y.; Wang, L.; Li, Z.; Zhang, X.H.; Yu, M. Bacterial community associated with healthy and diseased Pacific white shrimp (Litopenaeus vannamei) larvae and rearing water across different growth stages. Front. Microbiol. 2017, 8, 1362. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Fouhy, F.; Sleator, R.D.; O’ Driscoll, A.; Stanton, C.; Cotter, P.D.; Claesson, M.J. Comparing Apples and Oranges? Next Generation Sequencing and Its Impact on Microbiome Analysis. PLoS ONE 2016, 5, e0148028. [Google Scholar] [CrossRef] [PubMed]
- Naqib, A.; Jeon, T.; Kunstman, K.; Wang, W.; Shen, Y.; Sweeney, D.; Hyde, M.; Green, S.J. PCR effects of melting temperature adjustment of individual primers in degenerate primer pools. PeerJ 2019, 4, e6570. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-López, R.; Cornejo-Granados, F.; Lopez-Zavala, A.A.; Sánchez-López, F.; Cota-Huízar, A.; Sotelo-Mundo, R.R.; Guerrero, A.; Mendoza-Vargas, A.; Gómez-Gil, B.; Ochoa-Leyva, A. Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms 2020, 8, 134. https://doi.org/10.3390/microorganisms8010134
García-López R, Cornejo-Granados F, Lopez-Zavala AA, Sánchez-López F, Cota-Huízar A, Sotelo-Mundo RR, Guerrero A, Mendoza-Vargas A, Gómez-Gil B, Ochoa-Leyva A. Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms. 2020; 8(1):134. https://doi.org/10.3390/microorganisms8010134
Chicago/Turabian StyleGarcía-López, Rodrigo, Fernanda Cornejo-Granados, Alonso A. Lopez-Zavala, Filiberto Sánchez-López, Andrés Cota-Huízar, Rogerio R. Sotelo-Mundo, Abraham Guerrero, Alfredo Mendoza-Vargas, Bruno Gómez-Gil, and Adrian Ochoa-Leyva. 2020. "Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota" Microorganisms 8, no. 1: 134. https://doi.org/10.3390/microorganisms8010134
APA StyleGarcía-López, R., Cornejo-Granados, F., Lopez-Zavala, A. A., Sánchez-López, F., Cota-Huízar, A., Sotelo-Mundo, R. R., Guerrero, A., Mendoza-Vargas, A., Gómez-Gil, B., & Ochoa-Leyva, A. (2020). Doing More with Less: A Comparison of 16S Hypervariable Regions in Search of Defining the Shrimp Microbiota. Microorganisms, 8(1), 134. https://doi.org/10.3390/microorganisms8010134