Genome Analyses and Genome-Centered Metatranscriptomics of Methanothermobacter wolfeii Strain SIV6, Isolated from a Thermophilic Production-Scale Biogas Fermenter


Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of the Strain M. wolfeii SIV6 from the Thermophilic Biogas Fermenter
2.2. Sequencing, Assembly and Annotation of the M. wolfeii SIV6 Genome
2.3. Comparative Analyses of Methanothermobacter Genome Sequences
2.4. Metatranscriptome Mapping
2.5. Metagenome Fragment Recruitment
3. Results and Discussion
3.1. General Genome Features of M. wolfeii Strain SIV6
3.2. Phylogenetic Classification as Deduced from Comparative Genome Analyses
3.3. Genome Features of M. wolfeii SIV6 in Combination with In Situ Genome-Centered Metatranscriptomics
3.3.1. Functional Genome Annotation in Combination with Transcriptional Activity of Genes
3.3.2. Strain Specific Genome Features as Deduced from Singleton Analyses
3.3.3. Genomic Islands and Restriction-Modification Systems as Additional Genome Features of M. wolfeii SIV6
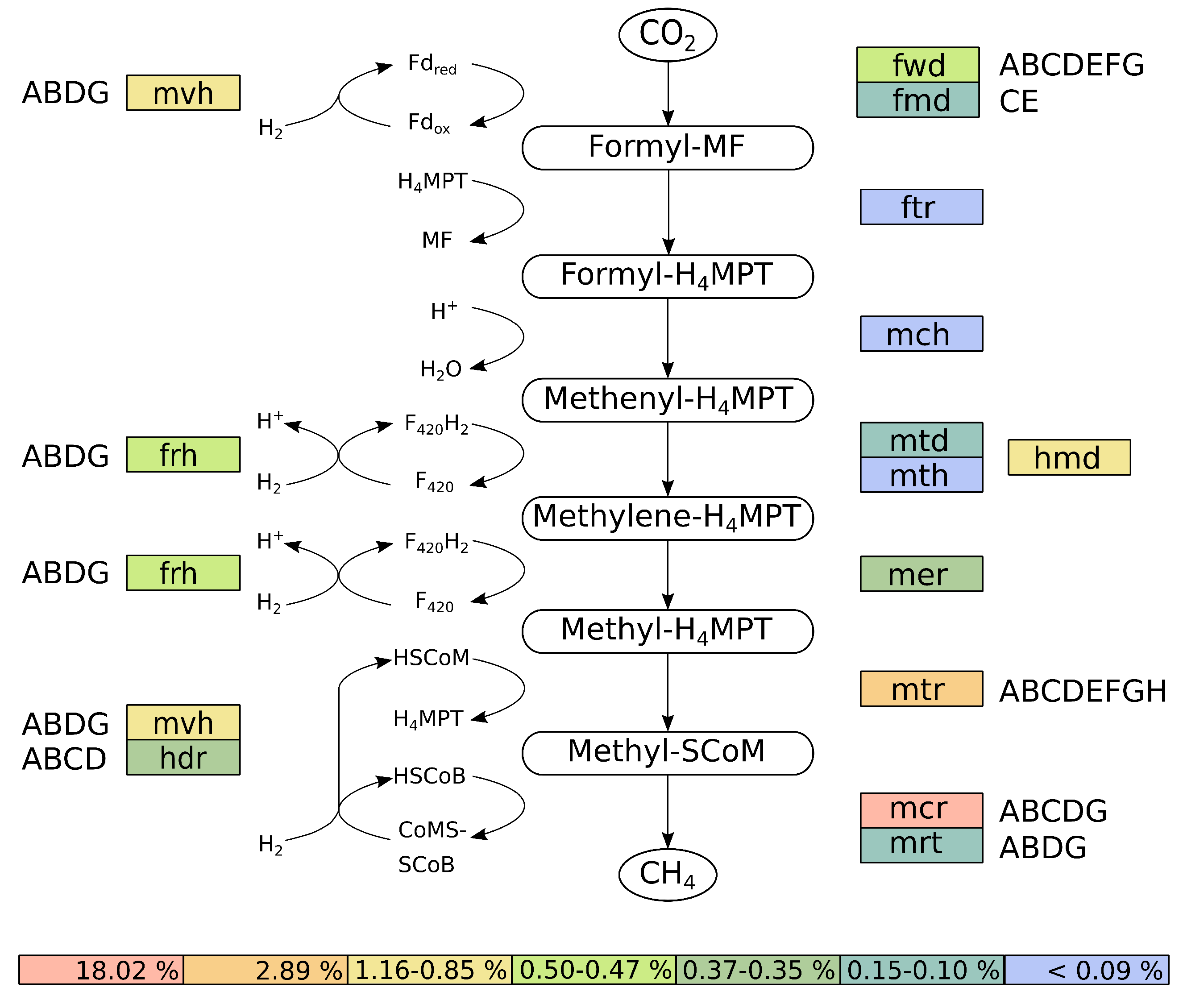
3.3.4. Reconstruction and Transcriptional Activity of the Hydrogenotrophic Methanogenesis Pathway of M. wolfeii SIV6
3.4. Fragment Recruitment of Metagenomic Reads from the Corresponding Thermophilic Biogas Fermenter on the M. wolfeii SIV6 Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zinder, S.H. Physiological ecology of methanogens. In Methanogenesis; Springer: Berlin/Heidelberg, Germany, 1993; pp. 128–206. [Google Scholar]
- Conrad, R. Contribution of hydrogen to methane production and control of hydrogen concentrations in methanogenic soils and sediments. FEMS Microbiol. Ecol. 1999, 28, 193–202. [Google Scholar] [CrossRef]
- Wirth, R.; Kovács, E.; Maróti, G.; Bagi, Z.; Rákhely, G.; Kovács, K.L. Characterization of a biogas-producing microbial community by short-read next generation DNA sequencing. Biotechnol. Biofuels 2012, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Stolze, Y.; Bremges, A.; Rumming, M.; Henke, C.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production-scale biogas plants. Biotechnol. Biofuels 2016, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 2019, 17, 219. [Google Scholar] [CrossRef]
- Ferrer, P.; Cambra-López, M.; Cerisuelo, A.; Peñaranda, D.S.; Moset, V. The use of agricultural substrates to improve methane yield in anaerobic co-digestion with pig slurry: Effect of substrate type and inclusion level. Waste Manag. 2014, 34, 196–203. [Google Scholar] [CrossRef]
- Ziganshin, A.M.; Liebetrau, J.; Pröter, J.; Kleinsteuber, S. Microbial community structure and dynamics during anaerobic digestion of various agricultural waste materials. Appl. Microbiol. Biotechnol. 2013, 97, 5161–5174. [Google Scholar] [CrossRef]
- Maus, I.; Koeck, D.E.; Cibis, K.G.; Hahnke, S.; Kim, Y.S.; Langer, T.; Kreubel, J.; Erhard, M.; Bremges, A.; Off, S.; et al. Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol. Biofuels 2016, 9, 171. [Google Scholar] [CrossRef]
- Yu, D.; Kurola, J.; Lähde, K.; Kymäläinen, M.; Sinkkonen, A.; Romantschuk, M. Biogas production and methanogenic archaeal community in mesophilic and thermophilic anaerobic co-digestion processes. J. Environ. Manag. 2014, 143, 54–60. [Google Scholar] [CrossRef]
- Hassa, J.; Maus, I.; Off, S.; Pühler, A.; Scherer, P.; Klocke, M.; Schlüter, A. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl. Microbiol. Biotechnol. 2018, 102, 5045–5063. [Google Scholar] [CrossRef]
- Rabii, A.; Aldin, S.; Dahman, Y.; Elbeshbishy, E. A review on anaerobic co-digestion with a focus on the microbial populations and the effect of multi-stage digester configuration. Energies 2019, 12, 1106. [Google Scholar] [CrossRef]
- Parte, A.C. LPSN–List of Prokaryotic names with Standing in Nomenclature (bacterio. net), 20 years on. Int. J. Syst. Evol. Microbiol. 2018, 68, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Dai, L.; Li, X.; Zhang, H.; Lu, Y. Isolation and characterization of Methanothermobacter crinale sp. nov., a novel hydrogenotrophic methanogen from the Shengli oil field. Appl. Environ. Microbiol. 2011, 77, 5212–5219. [Google Scholar] [CrossRef] [PubMed]
- Kotelnikova, S.; Obraztsova, A.Y.; Gongadze, G.; Laurinavichius, K. Methanobacterium thermoflexum sp. nov. and Methanobacterium defluvii sp. nov., thermophilic rod-shaped methanogens isolated from anaerobic digestor sludge. Syst. Appl. Microbiol. 1993, 16, 427–435. [Google Scholar] [CrossRef]
- Liesegang, H.; Kaster, A.K.; Wiezer, A.; Goenrich, M.; Wollherr, A.; Seedorf, H.; Gottschalk, G.; Thauer, R.K. Complete genome sequence of Methanothermobacter marburgensis, a methanoarchaeon model organism. J. Bacteriol. 2010, 192, 5850–5851. [Google Scholar] [CrossRef] [PubMed]
- Zeikus, J.; Wolfe, R. Methanobacterium thermoautotrophicus should be Methanobacterium thermoautotrophicum. Int. J. Syst. Evol. Microbiol. 1972, 22, 395. [Google Scholar] [CrossRef]
- Laurinavichus, K.S.; Kotelnikova, S.V.; Obraztsova, A.Y. A new species of the thermophilic methane-forming bacterium Methanobacterium thermophilum. Mikrobiologiya 1988, 57, 1035–1041. [Google Scholar]
- Nakamura, K.; Takahashi, A.; Mori, C.; Tamaki, H.; Mochimaru, H.; Nakamura, K.; Takamizawa, K.; Kamagata, Y. Methanothermobactertenebrarum sp. nov., a hydrogenotrophic, thermophilic methanogen isolated from gas-associated formation water of a natural gas field. Int. J. Syst. Evol. Microbiol. 2013, 63, 715–722. [Google Scholar] [CrossRef]
- Winter, J.; Lerp, C.; Zabel, H.P.; Wildenauer, F.; König, H.; Schindler, F. Methanobacterium wolfei, sp. nov., a new tungsten-requiring, thermophilic, autotrophic methanogen. Syst. Appl. Microbiol. 1984, 5, 457–466. [Google Scholar] [CrossRef]
- Kosaka, T.; Toh, H.; Toyoda, A. Complete genome sequence of a thermophilic hydrogenotrophic methanogen, Methanothermobacter sp. strain CaT2. Genome Announc. 2013, 1, e00672-13. [Google Scholar] [CrossRef]
- Smith, D.R.; Doucette-Stamm, L.A.; Deloughery, C.; Lee, H.; Dubois, J.; Aldredge, T.; Bashirzadeh, R.; Blakely, D.; Cook, R.; Gilbert, K.; et al. Complete genome sequence of Methanobacterium thermoautotrophicum deltaH: Functional analysis and comparative genomics. J. Bacteriol. 1997, 179, 7135–7155. [Google Scholar] [CrossRef]
- Balch, W.; Fox, G.; Magrum, L.; Woese, C.; Wolfe, R. Methanogens: Reevaluation of a unique biological group. Microbiol. Rev. 1979, 43, 260. [Google Scholar]
- Gordon, D.; Abajian, C.; Green, P. Consed: A graphical tool for sequence finishing. Genome Res. 1998, 8, 195–202. [Google Scholar] [CrossRef]
- Wibberg, D.; Blom, J.; Jaenicke, S.; Kollin, F.; Rupp, O.; Scharf, B.; Schneiker-Bekel, S.; Sczcepanowski, R.; Goesmann, A.; Setubal, J.C.; et al. Complete genome sequencing of Agrobacterium sp. H13-3, the former Rhizobium lupini H13-3, reveals a tripartite genome consisting of a circular and a linear chromosome and an accessory plasmid but lacking a tumor-inducing Ti-plasmid. J. Biotechnol. 2011, 155, 50–62. [Google Scholar] [CrossRef]
- Meyer, F.; Goesmann, A.; McHardy, A.C.; Bartels, D.; Bekel, T.; Clausen, J.; Kalinowski, J.; Linke, B.; Rupp, O.; Giegerich, R.; et al. GenDB—An open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 2003, 31, 2187–2195. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.; Simon Fraser University Research Computing Group. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, D566–D573. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, Z.; Fu, L.; Niu, B.; Li, W. WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011, 12, 444. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Blom, J.; Kreis, J.; Spänig, S.; Juhre, T.; Bertelli, C.; Ernst, C.; Goesmann, A. EDGAR 2.0: An enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 2016, 44, W22–W28. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Targeted in situ metatranscriptomics for selected taxa from mesophilic and thermophilic biogas plants. Microb. Biotechnol. 2018, 11, 667–679. [Google Scholar] [CrossRef]
- Reddy, T.B.; Thomas, A.D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E.A.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v. 5: A metadata management system based on a four level (meta) genome project classification. Nucleic Acids Res. 2014, 43, D1099–D1106. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Hilker, R.; Stadermann, K.B.; Schwengers, O.; Anisiforov, E.; Jaenicke, S.; Weisshaar, B.; Zimmermann, T.; Goesmann, A. ReadXplorer 2—Detailed read mapping analysis and visualization from one single source. Bioinformatics 2016, 32, 3702–3708. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Schlüter, A.; Bekel, T.; Diaz, N.N.; Dondrup, M.; Eichenlaub, R.; Gartemann, K.H.; Krahn, I.; Krause, L.; Krömeke, H.; Kruse, O.; et al. The metagenome of a biogas-producing microbial community of a production-scale biogas plant fermenter analysed by the 454-pyrosequencing technology. J. Biotechnol. 2008, 136, 77–90. [Google Scholar] [CrossRef]
- Eikmeyer, F.G.; Köfinger, P.; Poschenel, A.; Jünemann, S.; Zakrzewski, M.; Heinl, S.; Mayrhuber, E.; Grabherr, R.; Pühler, A.; Schwab, H.; et al. Metagenome analyses reveal the influence of the inoculant Lactobacillus buchneri CD034 on the microbial community involved in grass ensiling. J. Biotechnol. 2013, 167, 334–343. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Figueras, M.J.; Beaz-Hidalgo, R.; Hossain, M.J.; Liles, M.R. Taxonomic affiliation of new genomes should be verified using average nucleotide identity and multilocus phylogenetic analysis. Genome Announc. 2014, 2, e00927-14. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Goesmann, A.; Jaenicke, S.; Jünemann, S.; Eikmeyer, F.; Szczepanowski, R.; Al-Soud, W.A.; Sørensen, S.; Pühler, A.; Schlüter, A. Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J. Biotechnol. 2012, 158, 248–258. [Google Scholar] [CrossRef]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schlüter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Pühler, A.; Reichl, U.; Klocke, M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst. Appl. Microbiol. 2013, 36, 330–338. [Google Scholar] [CrossRef]
- Ortseifen, V.; Stolze, Y.; Maus, I.; Sczyrba, A.; Bremges, A.; Albaum, S.P.; Jaenicke, S.; Fracowiak, J.; Pühler, A.; Schlüter, A. An integrated metagenome and-proteome analysis of the microbial community residing in a biogas production plant. J. Biotechnol. 2016, 231, 268–279. [Google Scholar] [CrossRef]
- Heyer, R.; Benndorf, D.; Kohrs, F.; De Vrieze, J.; Boon, N.; Hoffmann, M.; Rapp, E.; Schlüter, A.; Sczyrba, A.; Reichl, U. Proteotyping of biogas plant microbiomes separates biogas plants according to process temperature and reactor type. Biotechnol. Biofuels 2016, 9, 155. [Google Scholar] [CrossRef]
- Benaroudj, N.; Goldberg, A.L. PAN, the proteasome-activating nucleotidase from archaebacteria, is a protein-unfolding molecular chaperone. Nat. Cell Biol. 2000, 2, 833. [Google Scholar] [CrossRef]
- Meyer, B.H.; Albers, S.V. Hot and sweet: Protein glycosylation in Crenarchaeota. Biochem. Soc. Trans. 2013, 41, 384–392. [Google Scholar] [CrossRef]
- Calo, D.; Kaminski, L.; Eichler, J. Protein glycosylation in Archaea: Sweet and extreme. Glycobiology 2010, 20, 1065–1076. [Google Scholar] [CrossRef]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006, 16, 29R–37R. [Google Scholar] [CrossRef]
- Rodrigues-Oliveira, T.; Belmok, A.; Vasconcellos, D.; Schuster, B.; Kyaw, C.M. Archaeal S-layers: Overview and current state of the art. Front. Microbiol. 2017, 8, 2597. [Google Scholar] [CrossRef]
- Lu, D.; Yang, C.; Liu, Z. How hydrophobicity and the glycosylation site of glycans affect protein folding and stability: A molecular dynamics simulation. J. Phys. Chem. B 2011, 116, 390–400. [Google Scholar] [CrossRef]
- Peyfoon, E.; Meyer, B.; Hitchen, P.G.; Panico, M.; Morris, H.R.; Haslam, S.M.; Albers, S.V.; Dell, A. The S-layer glycoprotein of the crenarchaeote Sulfolobus acidocaldarius is glycosylated at multiple sites with chitobiose-linked N-glycans. Archaea 2010, 2010, 754101. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, Y. Not all predicted CRISPR–Cas systems are equal: Isolated cas genes and classes of CRISPR like elements. BMC Bioinform. 2017, 18, 92. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Q.; Zhang, Q.; Wang, T.; Yue, H.; Wu, L.; Shi, J.; Qin, Z.; Zhou, J.; Zuo, J.; et al. Bacteriophage–prokaryote dynamics and interaction within anaerobic digestion processes across time and space. Microbiome 2017, 5, 57. [Google Scholar] [CrossRef]
- Schlesner, M.; Miller, A.; Streif, S.; Staudinger, W.F.; Müller, J.; Scheffer, B.; Siedler, F.; Oesterhelt, D. Identification of Archaea-specific chemotaxis proteins which interact with the flagellar apparatus. BMC Microbiol. 2009, 9, 56. [Google Scholar] [CrossRef]
- Albers, S.V.; Jarrell, K.F. The archaellum: An update on the unique archaeal motility structure. Trends Microbiol. 2018, 26, 351–362. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Towards functional characterization of archaeal genomic dark matter. Biochem. Soc. Trans. 2019, 47, 389. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Detecting genomic islands using bioinformatics approaches. Nat. Rev. Microbiol. 2010, 8, 373. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2014, 43, D298–D299. [Google Scholar] [CrossRef]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef]
- Fullmer, M.S.; Ouellette, M.; Louyakis, A.S.; Papke, R.T.; Gogarten, J.P. The Patchy Distribution of Restriction–Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria. Genes 2019, 10, 233. [Google Scholar] [CrossRef]
- Ershova, A.; Rusinov, I.; Spirin, S.; Karyagina, A.; Alexeevski, A. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry 2015, 80, 1373–1386. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Siewert, C.; Kohrs, F.; Greve, J.; Maus, I.; Klang, J.; Klocke, M.; Heiermann, M.; Hoffmann, M.; et al. Metaproteome analysis reveals that syntrophy, competition, and phage-host interaction shape microbial communities in biogas plants. Microbiome 2019, 7, 69. [Google Scholar] [CrossRef]
- Reeve, J.N.; Nölling, J.; Morgan, R.M.; Smith, D.R. Methanogenesis: Genes, genomes, and who’s on first? J. Bacteriol. 1997, 179, 5975. [Google Scholar] [CrossRef]
- Cedervall, P.E.; Dey, M.; Pearson, A.R.; Ragsdale, S.W.; Wilmot, C.M. Structural insight into methyl-coenzyme M reductase chemistry using coenzyme B analogues. Biochemistry 2010, 49, 7683–7693. [Google Scholar] [CrossRef]
- Buckel, W.; Thauer, R.K. Flavin-based electron bifurcation, a new mechanism of biological energy coupling. Chem. Rev. 2018, 118, 3862–3886. [Google Scholar] [CrossRef]
- Kougias, P.G.; Campanaro, S.; Treu, L.; Zhu, X.; Angelidaki, I. A novel archaeal species belonging to Methanoculleus genus identified via de-novo assembly and metagenomic binning process in biogas reactors. Anaerobe 2017, 46, 23–32. [Google Scholar] [CrossRef]
- Cadillo-Quiroz, H. Contribution of transcriptomics to systems-level understanding of methanogenic archaea. Archaea 2013, 2013, 586369. [Google Scholar]
- Hendrickson, E.L.; Haydock, A.K.; Moore, B.C.; Whitman, W.B.; Leigh, J.A. Functionally distinct genes regulated by hydrogen limitation and growth rate in methanogenic Archaea. Proc. Natl. Acad. Sci. USA 2007, 104, 8930–8934. [Google Scholar] [CrossRef]
- Xia, Q.; Wang, T.; Hendrickson, E.L.; Lie, T.J.; Hackett, M.; Leigh, J.A. Quantitative proteomics of nutrient limitation in the hydrogenotrophic methanogen Methanococcus maripaludis. BMC Microbiol. 2009, 9, 149. [Google Scholar] [CrossRef]
- Westerholm, M.; Müller, B.; Singh, A.; Karlsson Lindsjö, O.; Schnürer, A. Detection of novel syntrophic acetate-oxidizing bacteria from biogas processes by continuous acetate enrichment approaches. Microb. Biotechnol. 2018, 11, 680–693. [Google Scholar] [CrossRef]
- Theuerl, S.; Klang, J.; Prochnow, A. Process disturbances in agricultural biogas production—Causes, mechanisms and effects on the biogas microbiome: A review. Energies 2019, 12, 365. [Google Scholar] [CrossRef]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol. Mol. Biol. Rev. 1997, 61, 262–280. [Google Scholar]
- Luo, H.W.; Zhang, H.; Suzuki, T.; Hattori, S.; Kamagata, Y. Differential expression of methanogenesis genes of Methanothermobacter thermoautotrophicus (formerly Methanobacterium thermoautotrophicum) in pure culture and in cocultures with fatty acid-oxidizing syntrophs. Appl. Environ. Microbiol. 2002, 68, 1173–1179. [Google Scholar] [CrossRef]
- Theuerl, S.; Herrmann, C.; Heiermann, M.; Grundmann, P.; Landwehr, N.; Kreidenweis, U.; Prochnow, A. The future agricultural biogas plant in Germany: A vision. Energies 2019, 12, 396. [Google Scholar] [CrossRef]
- Zakaria, B.S.; Dhar, B.R. Progress towards catalyzing electro-methanogenesis in anaerobic digestion process: Fundamentals, process optimization, design and scale-up considerations. Bioresource Technol. 2019, 289, 121738. [Google Scholar] [CrossRef]
- Mayer, F.; Enzmann, F.; Lopez, A.M.; Holtmann, D. Performance of different methanogenic species for the microbial electrosynthesis of methane from carbon dioxide. Bioresource Technol. 2019, 289, 121706. [Google Scholar] [CrossRef]
- Blasco-Gómez, R.; Batlle-Vilanova, P.; Villano, M.; Balaguer, M.D.; Colprim, J.; Puig, S. On the edge of research and technological application: A critical review of electromethanogenesis. Int. J. Mol. Sci. 2017, 18, 874. [Google Scholar] [CrossRef]
- Fu, Q.; Kuramochi, Y.; Fukushima, N.; Maeda, H.; Sato, K.; Kobayashi, H. Bioelectrochemical analyses of the development of a thermophilic biocathode catalyzing electromethanogenesis. Environ. Sci. Technol. 2015, 49, 1225–1232. [Google Scholar] [CrossRef]
- Kobayashi, H.; Sun, X.; Fu, Q.; Maeda, H.; Sato, K. Draft Genome Sequence of Methanothermobacter sp. Strain EMTCatA1, Reconstructed from the Metagenome of a Thermophilic Electromethanogenesis- Catalyzing Biocathode. Genome Announc. 2017, 5, e00892-17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Origin | Reference |
|---|---|---|---|
| M. crinale | Tm2 | Oil field | Cheng et al. (2011) [13] |
| M. defluvii | ADZ | Digester sludge | Kotelnikova et al. (1993) [14] |
| M. marburgensis | Marburg | Sewage sludge | Liesegang et al. (2010) [15] |
| M. thermautotrophicus | H | Sewage sludge | Zeikus and Wolfe (1972) [16] |
| M. thermoflexus | IDZ | Digester sludge | Kotelnikova et al. (1993) [14] |
| M. thermophilus | M | Thermophilic methane tank | Laurinavichus et al. (1988) [17] |
| M. tenebrarum | RMAS | Gas field | Nakamura et al. (2013) [18] |
| M. wolfeii | JCM 14652 | Sewage sludge | Winter et al. (1984) [19] |
| General Features | M. wolfeii SIV6 |
|---|---|
| Size (bp) | 1,686,891 |
| GC content (%) | 48.89 |
| Total genes | 1786 |
| Protein coding genes | 1659 |
| Genes assigned to COG categories | 1498 |
| rrn operons | 2 |
| tRNA genes | 36 |
| Genomic islands | 3 |
| Singleton Number | Locus | Gene Annotation | Predicted Function | GI | TPM |
|---|---|---|---|---|---|
| 1 | MWSIV6_ 0587 | Glycosyltransferase | protein glycosylation | - | 22 |
| 2 | MWSIV6_ 0588 | Glycosyltransferase | protein glycosylation | - | 20 |
| 3 | MWSIV6_ 0666 | 4Fe-4S ferredoxin | mediating the transfer of electrons | - | 10 |
| in different metabolic reactions | |||||
| 4 | MWSIV6_ 0722 | Hexosyltransferase | protein glycosylation | - | 520 |
| 5 | MWSIV6_ 0726 | Glycosyltransferase | protein glycosylation | - | 202 |
| 6 | MWSIV6_ 0728 | Thymidylate kinase | DNA synthesis | - | 29 |
| 7 | MWSIV6_ 0729 | Alkaline Phosphatase | post-translational modification | - | 47 |
| 8 | MWSIV6_ 0732 | Uncharacterized | protein glycosylation | - | 52 |
| Glycosyltransferase | |||||
| 9 | MWSIV6_ 0863 | ATPase | drives the transport of protons or | - | 79 |
| other cations across the cell membrane | |||||
| 10 | MWSIV6_ 0990 | Cl-channel voltage- | transfers chloride ions | - | 26 |
| gated family protein | across the membrane | ||||
| 11 | MWSIV6_ 1297 | PBS lyase HEAT | archaeal chemotaxis | GI 2 | 0 |
| domain protein | |||||
| 12 | MWSIV6_ 1305 | Integrase family protein | DNA breaking and rejoining | GI 2 | 2 |
| 13 | MWSIV6_ 1466 | CRISPR-associated | defense system | - | 33 |
| protein Csx1 | |||||
| 14 | MWSIV6_ 1483 | CRISPR-associated | defense system | GI 3 | 143 |
| nuclease/helicase Cas3 | |||||
| 15 | MWSIV6_ 1484 | CRISPR-associated | defense system | GI 3 | 126 |
| protein Cas5 | |||||
| 16 | MWSIV6_ 1485 | CRISPR-associated | defense system | GI 3 | 300 |
| protein Cas7 |
| Glycosyltransferase Family | 1 | 2 | 4 | 20 | 66 | 81 | NC | Total |
|---|---|---|---|---|---|---|---|---|
| M. wolfeii SIV6 | 1 | 7 | 13 | 1 | 5 | 1 | 1 | 29 |
| Methanothermobacter sp. CaT2 | 1 | 9 | 8 | 1 | 4 | 1 | 1 | 25 |
| M. thermautotrophicusH | 1 | 9 | 9 | 1 | 4 | 1 | 0 | 25 |
| M. marburgensis str. Marburg | 1 | 7 | 8 | 1 | 3 | 1 | 1 | 22 |
| Gene | Predicted product | TPM |
|---|---|---|
| mcrD | Methyl-coenzyme M reductase I operon protein D | 74,808 |
| mcrC | Methyl-coenzyme M reductase I operon protein C | 40,551 |
| mcrB | Methyl-coenzyme M reductase I subunit beta | 27,412 |
| mcrG | Methyl-coenzyme M reductase I subunit gamma | 21,785 |
| mcrA | Methyl-coenzyme M reductase I subunit alpha | 15,684 |
| hmd | 5,10-methylenetetrahydromethanopterin reductase | 11,581 |
| sod | DNA-directed RNA polymerase subunit HD | 8847 |
| mtrA1 | F420-non-reducing hydrogenase | 5997 |
| mtrF | Proteasome-activating nucleotidase | 5729 |
| mtrG | 50S ribosomal protein L29P | 4138 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassa, J.; Wibberg, D.; Maus, I.; Pühler, A.; Schlüter, A. Genome Analyses and Genome-Centered Metatranscriptomics of Methanothermobacter wolfeii Strain SIV6, Isolated from a Thermophilic Production-Scale Biogas Fermenter. Microorganisms 2020, 8, 13. https://doi.org/10.3390/microorganisms8010013
Hassa J, Wibberg D, Maus I, Pühler A, Schlüter A. Genome Analyses and Genome-Centered Metatranscriptomics of Methanothermobacter wolfeii Strain SIV6, Isolated from a Thermophilic Production-Scale Biogas Fermenter. Microorganisms. 2020; 8(1):13. https://doi.org/10.3390/microorganisms8010013
Chicago/Turabian StyleHassa, Julia, Daniel Wibberg, Irena Maus, Alfred Pühler, and Andreas Schlüter. 2020. "Genome Analyses and Genome-Centered Metatranscriptomics of Methanothermobacter wolfeii Strain SIV6, Isolated from a Thermophilic Production-Scale Biogas Fermenter" Microorganisms 8, no. 1: 13. https://doi.org/10.3390/microorganisms8010013
APA StyleHassa, J., Wibberg, D., Maus, I., Pühler, A., & Schlüter, A. (2020). Genome Analyses and Genome-Centered Metatranscriptomics of Methanothermobacter wolfeii Strain SIV6, Isolated from a Thermophilic Production-Scale Biogas Fermenter. Microorganisms, 8(1), 13. https://doi.org/10.3390/microorganisms8010013