Genome Insights into the Novel Species Microvirga brassicacearum, a Rapeseed Endophyte with Biotechnological Potential

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Isolation and Identification
2.2. DNA Extraction and 16S rRNA Amplification and Sequencing
2.3. Draft Genome Sequencing and Annotation
2.4. Genotypic Analysis of Microvirga brassicacearum CDVBN77T
2.5. Chemotaxonomic Analysis of Microvirga brassicacearum CDVBN77T
2.6. Microvirga brassicacearum CDVBN77T Bacterial Characterization
3. Results
3.1. Bacterial Isolation and Identification
3.2. Genome Properties and Comparison with Those of Its Closest Related Species
3.3. Plant Growth Promotion Potential
3.4. Potential for the Production of Enzymes with Biotechnological Potential
3.5. Genome Mining of Gene Clusters Associated to the Biosynthesis of Secondary Metabolites
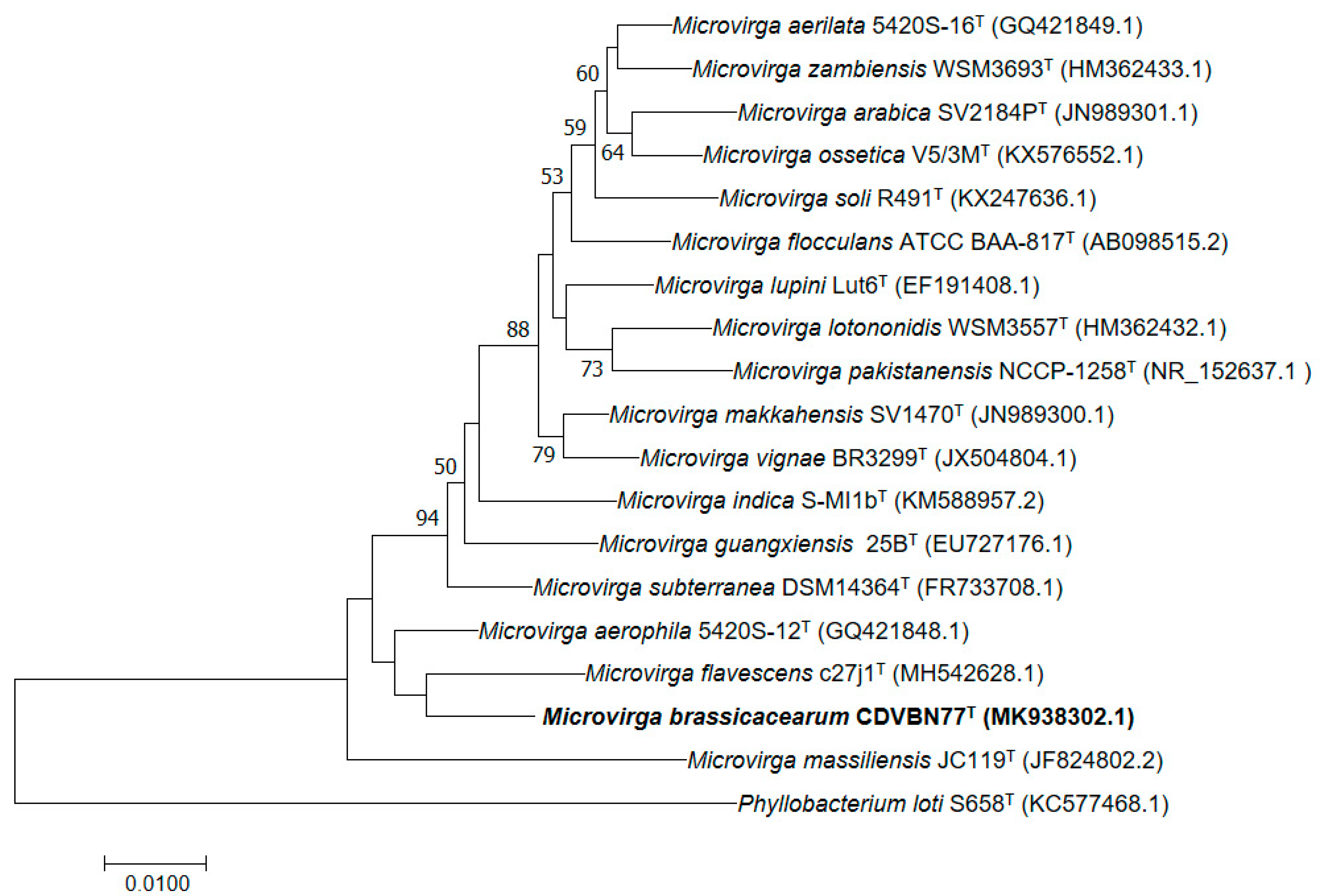
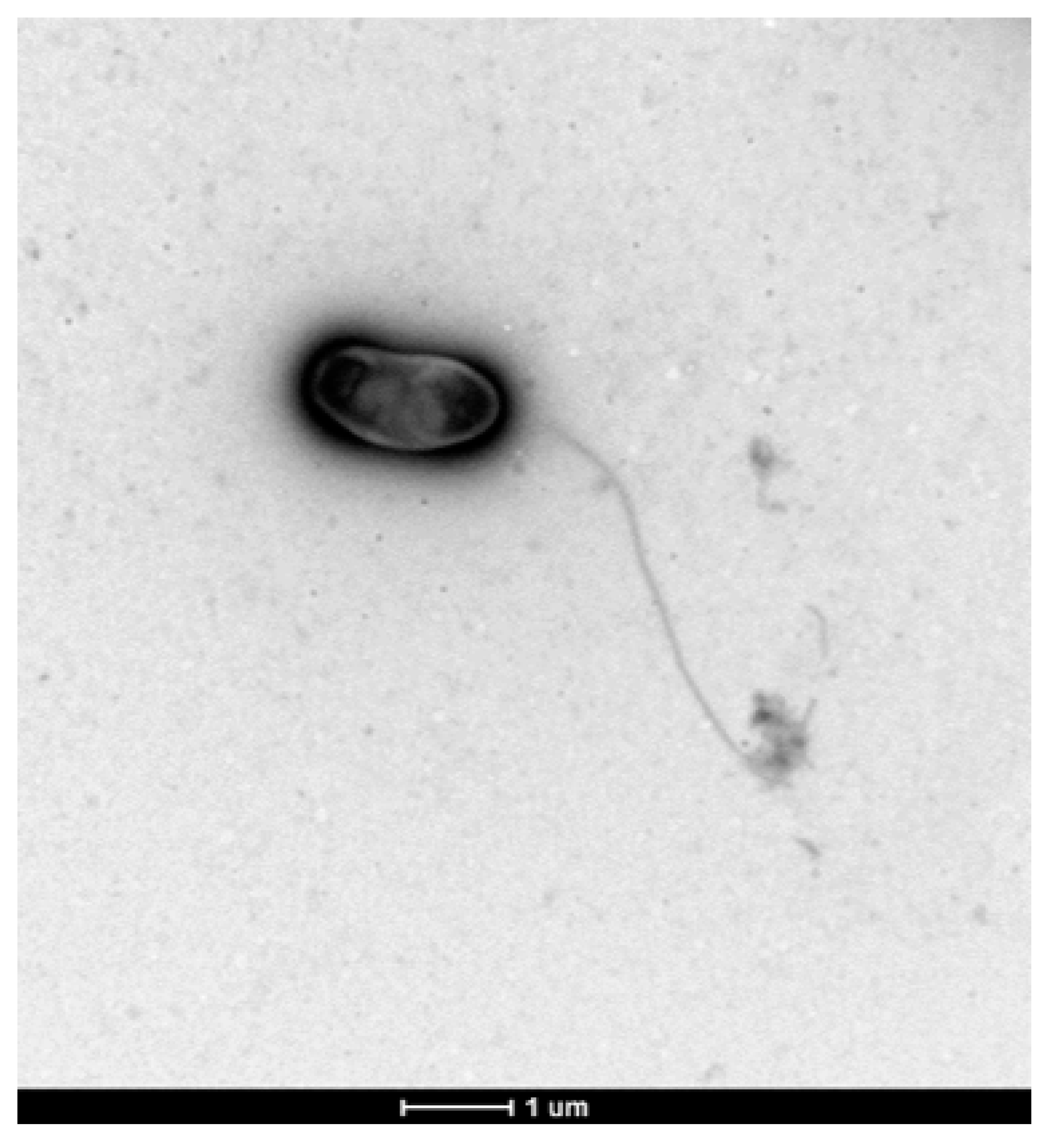
3.6. Colony and Cellular Morphology in Strain CDVBN77T
3.7. Phenotypic and Chemotaxonomic Characterization of Strain CDVBN77T
4. Discussion
5. Description of Microvirga brassicacearum sp. nov.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Locey, K.J.; Lennon, J.T. Scaling laws predict global microbial diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 5970–5975. [Google Scholar] [CrossRef] [PubMed]
- Jongsik, C.; Oren, A.; Ventosa, A.; Christensen, H.; Ruiz, D.; Da Costa, M.; Rooney, A.P.; Yi, H.; Wei, X.; de Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar]
- Auch, A.F.; von Jan, M.; Klenk, H.P.; Göker, M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genom. Sci. 2010, 2, 117–134. [Google Scholar]
- López-Mondéjar, R.; Kostovčík, M.; Lladó, S.; Carro, L.; García-Fraile, P. Exploring the Plant Microbiome Through Multi-omics Approaches. Probiotics Agroecosystem 2017, 233–268. [Google Scholar]
- Saati-Santamaría, Z.; López-Mondéjar, R.; Jiménez-Gómez, A.; Díez-Méndez, A.; Větrovský, T.; Igual, J.M.; Velázquez, E.; Kolarik, M.; Rivas, R.; García-Fraile, P. Discovery of phloeophagus beetles as a source of Pseudomonas strains that produce potentially new bioactive substances and description of Pseudomonas bohemica sp. nov. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Fabryová, A.; Kostovčík, M.; Díez-Méndez, A.; Jiménez-Gómez, A.; Celador-Lera, L.; Saati-Santamaría, Z.; Sechovcová, H.; Menéndez, E.; Kolarik, M.; García-Fraile, P. On the bright side of a forest pest-the metabolic potential of bark beetles’ bacterial associates. Sci. Total Environ. 2018, 619, 9–17. [Google Scholar] [CrossRef]
- Gopal, M.; Gupta, A. Microbiome selection could spur next-generation plant breeding strategies. Front. Microbiol. 2016, 7, 1971. [Google Scholar] [CrossRef]
- Menéndez, E.; Garcia-Fraile, P. Plant probiotic bacteria: Solutions to feed the world. Aims Microbiol. 2017, 3, 502–524. [Google Scholar] [CrossRef]
- Caputo, A.; Lagier, J.C.; Azza, S.; Robert, C.; Mouelhi, D.; Fournier, P.E.; Raoult, D. Microvirga massiliensis sp. nov., the human commensal with the largest genome. Microbiol. Open 2016, 5, 307–322. [Google Scholar] [CrossRef]
- Weon, H.Y.; Kwon, S.W.; Son, J.A.; Jo, E.H.; Kim, S.J.; Kim, Y.S.; Kim, B.Y.; Ka, J.O. Description of Microvirga aerophila sp. nov. and Microvirga aerilata sp. nov., isolated from air, reclassification of Balneimonas flocculans Takeda et al. 2004 as Microvirga flocculans comb. nov. and emended description of the genus Microvirga. Int. J. Syst. Evol. Microbiol. 2010, 60, 2596–2600. [Google Scholar] [CrossRef][Green Version]
- Kanso, S.; Patel, B.K. Microvirga subterranea gen. nov., sp. nov., a moderate thermophile from a deep subsurface Australian thermal aquifer. Int. J. Syst. Evol. Microbiol. 2003, 53, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, F.; Xin, Y.H.; Zhang, J.; Fang, C. Microvirga guangxiensis sp. nov., a novel alphaproteobacterium from soil, and emended description of the genus Microvirga. Int. J. Syst. Evol. Microbiol. 2009, 59, 1997–2001. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amin, A.; Ahmed, I.; Habib, N.; Abbas, S.; Hasan, F.; Xiao, M.; Hozzein, W.N.; Li, W.J. Microvirga pakistanensis sp. nov., a novel bacterium isolated from desert soil of Cholistan, Pakistan. Arch. Microbiol. 2016, 198, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Dahal, R.H.; Kim, J. Microvirga soli sp. nov., an alphaproteobacterium isolated from soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 127–132. [Google Scholar] [PubMed]
- Tapase, S.R.; Mawlankar, R.B.; Sundharam, S.S.; Krishnamurthi, S.; Dastager, S.G.; Kodam, K.M. Microvirga indica sp. nov., an arsenite-oxidizing Alphaproteobacterium, isolated from metal industry waste soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Veyisoglu, A.; Tatar, D.; Saygin, H.; Inan, K.; Cetin, D.; Guven, K.; Tuncer, M.; Sahin, N. Microvirga makkahensis sp. nov., and Microvirga arabica sp. nov., isolated from sandy arid soil. Antonie Van Leeuwenhoek 2016, 109, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Zhang, J.; Yao, Q.; Zhu, H.H. Microvirga flavescens sp. nov., a novel bacterium isolated from forest soil and emended description of the genus Microvirga. Int. J. Syst. Evol. Microbiol. 2019, 69, 667–671. [Google Scholar] [CrossRef]
- Ardley, J.K.; Parker, M.A.; De Meyer, S.E.; Trengove, R.D.; O’Hara, G.W.; Reeve, W.G.; Yates, R.J.; Dilworth, M.J.; Willems, A.; Howieson, J.G. Microvirga lupini sp. nov., Microvirga lotononidis sp. nov., and Microvirga zambiensis sp. nov. are Alphaproteobacterial root nodule bacteria that specifically nodulate and fix nitrogen with geographically and taxonomically separate legume hosts. Int. J. Syst. Evol. Microbiol. 2012, 62, 2579–2588. [Google Scholar] [CrossRef]
- Safronova, V.I.; Kuznetsova, I.G.; Sazanova, A.L.; Belimov, A.A.; Andronov, E.E.; Chirak, E.R.; Osledkin, Y.S.; Onishchuk, O.P.; Kurchak, O.N.; Shaposhnikov, A.I.; et al. Microvirga ossetica sp. nov., a species of rhizobia isolated from root nodules of the legume species Vicia alpestris Steven. Int. J. Syst. Evol. Microbiol. 2017, 67, 94–100. [Google Scholar]
- Radl, V.; Simões-Araújo, J.L.; Leite, J.; Passos, S.R.; Martins, L.M.V.; Xavier, G.R.; Rumjanek, N.G.; Baldani, J.I.; Zilli, J.E. Microvirga vignae sp. nov., a root nodule symbiotic bacterium isolated from cowpea grown in semi-arid Brazil. Int. J. Syst. Evol. Microbiol. 2017, 64, 725–730. [Google Scholar] [CrossRef]
- Samrot, A.V.; Rio, A.J.; Kumar, S.S.; Samanvitha, S.K. Bioprospecting studies of pigmenting Pseudomonas aeruginosa SU-1, Microvirga aerilata SU14 and Bacillus megaterium SU15 isolated from garden soil. Biocatal. Agric. Biotechnol. 2017, 11, 330–337. [Google Scholar] [CrossRef]
- Jiménez-Gómez, A.; Saati-Santamaría, Z.; Menéndez, E.; Rivas, R.; Mateos, P.F.; Velázquez, E.; García-Fraile, P. Analysis of the biodiversity of Brassica napus bacterial endophytes and selection of potential efficient biofertilizers for rapeseed crops. under review. Front. Microbiol.
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Lee, J.H.; Jung, Y.; Kim, M.; Kim, S.; Kim, B.K.; Lim, Y.W. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2259–2261. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Meyer, F. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Yin, Y. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, 95–101. [Google Scholar]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, 81–87. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2017, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Mol. Biol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Menéndez, E.; Ramírez-Bahena, M.H.; Fabryová, A.; Igual, J.M.; Benada, O.; Mateos, P.F.; Peix, A.; Kolařík, M.; García-Fraile, P. Pseudomonas coleopterorum sp. nov., a cellulase-producing bacterium isolated from the bark beetle Hylesinus fraxini. Int. J. Syst. Evol. Microbiol. 2015, 65, 2852–2858. [Google Scholar]
- Lladó, S.; Benada, O.; Cajthaml, T.; Baldrian, P.; García-Fraile, P. Silvibacterium bohemicum gen. nov. sp. nov., an acidobacterium isolated from coniferous soil in the Bohemian Forest National Park. Syst. Appl. Microbiol. 2016, 39, 14–19. [Google Scholar]
- Doetsch, R.N. Determinative methods of light microscopy. Man. Meth. Gen. Bact. 1981, 21–33. [Google Scholar]
- Schwyn, B.; Neilands, J.B. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987, 160, 47–56. [Google Scholar] [CrossRef]
- Alexander, D.B.; Zuberer, D.A. Use of Chrome Azurol S reagents to evaluate siderophore production by rhizosphere bacteria. Biol. Fertil. Soils 1991, 12, 39–45. [Google Scholar] [CrossRef]
- Pikovskaya, R.I. Mobilization of phosphorus in soil connection with the vital activity of some microbial species. Microbiologiya 1948, 17, 362–370. [Google Scholar]
- Robledo, M.; Rivera, L.; Jiménez-Zurdo, J.I.; Rivas, R.; Dazzo, F.; Velázquez, E.; Martínez-Molina, E.; Hirsch, A.; Mateos, P.F. Role of Rhizobium endoglucanase CelC2 in cellulose biosynthesis and biofilm formation on plant roots and abiotic surfaces. Microb. Cell Fact. 2012, 11, 125. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.; Jung, B.K.; Shin, J.H. Complete genome sequence of Pseudomonas rhizosphaerae IH5T (= DSM 16299T), a phosphate-solubilizing rhizobacterium for bacterial biofertilizer. J. Biotechnol. 2015, 193, 137–138. [Google Scholar] [CrossRef] [PubMed]
- de Werra, P.; Péchy-Tarr, M.; Keel, C.; Maurhofer, M. Role of gluconic acid production in the regulation of biocontrol traits of Pseudomonas fluorescens CHA0. Appl. Environ. Microbiol. 2009, 75, 41624174. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.H. Involvement of the quinoprotein glucose dehydrogenase in the solubilization of exogenous phosphates by gram-negative bacteria. In Phosphate in microorganisms: Cellular and Molecular biology; ASM Press: Washington, DC, USA, 1994; pp. 197–203. [Google Scholar]
- Etesami, H.; Emami, S.; Alikhani, H.A. Potassium solubilizing bacteria (KSB): Mechanisms, promotion of plant growth, and future prospects A review. J. Soil Sci. Plant Nutr. 2017, 17, 897–911. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Hou, J.; Tu, C.; Luo, Y.; Christie, P. Whole genome analysis of halotolerant and alkalotolerant plant growth-promoting rhizobacterium Klebsiella sp. D5A. Sci. Rep. 2016, 6, 26710. [Google Scholar] [CrossRef]
- Epstein, W. The roles and regulation of potassium in bacteria. Prog. Nucleic Acid Res. 2003, 75, 293–320. [Google Scholar]
- Velázquez, E.; García-Fraile, P.; Ramírez-Bahena, M.H.; Rivas, R.; Martínez-Molina, E. Bacteria involved in nitrogen-fixing legume symbiosis: Current taxonomic perspective. Microbes Legume Improv. 2010, 1–25. [Google Scholar]
- Mateos, P.F.; Jimenez-Zurdo, J.I.; Chen, J.; Squartini, A.S.; Haack, S.K.; Martinez-Molina, E.; Hubbell, D.H.; Dazzo, F.B. Cell-associated pectinolytic and cellulolytic enzymes in Rhizobium leguminosarum biovar trifolii. Appl. Environ. Microbiol. 1992, 58, 1816–1822. [Google Scholar]
- Reed, J.W.; Walker, G.C. The exoD gene of Rhizobium meliloti encodes a novel function needed for alfalfa nodule invasion. J. Bacteriol. 1991, 173, 664–677. [Google Scholar] [CrossRef]
- Polissi, A.; Sperandeo, P. The lipopolysaccharide export pathway in Escherichia coli: Structure, organization and regulated assembly of the Lpt machinery. Mar. Drugs 2014, 12, 1023–1042. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, Z.; Tang, X.; Paterson, N.G.; Dong, C. Structural and functional insights into the lipopolysaccharide ABC transporter LptB 2 FG. Nat. Commun. 2017, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Pradel, E.; Parker, C.T.; Schnaitman, C.A. Structures of the rfaB, rfaI, rfaJ, and rfaS genes of Escherichia coli K-12 and their roles in assembly of the lipopolysaccharide core. J. Bacteriol. 1992, 174, 4736–4745. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, C.; Greenberg, E.P. Signalling: Listening in on bacteria: Acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 685. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Xu, F.; Zeng, J.; Zhan, J. Type III polyketide synthases in natural product biosynthesis. Iubmb Life 2012, 64, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Rivas, R.; García-Fraile, P.; Mateos, P.F.; Martínez-Molina, E.; Velazquez, E. Paenibacillus cellulosilyticus sp. nov., a cellulolytic and xylanolytic bacterium isolated from the bract phyllosphere of Phoenix dactylifera. Int. J. Syst. Evol. Microbiol. 2006, 56, 2777–2781. [Google Scholar] [CrossRef] [PubMed]
- Rivas, R.; Garcia-Fraile, P.; Zurdo-Pineiro, J.L.; Mateos, P.F.; Martinez-Molina, E.; Bedmar, E.J.; Sánchez-Raya, J.; Velazquez, E. Saccharibacillus sacchari gen. nov., sp. nov., isolated from sugar cane. Int. J. Syst. Evol. Microbiol. 2008, 58, 1850–1854. [Google Scholar] [CrossRef]
- García-Fraile, P.; Velázquez, E.; Mateos, P.F.; Martínez-Molina, E.; Rivas, R. Cohnella phaseoli sp. nov., isolated from root nodules of Phaseolus coccineus in Spain, and emended description of the genus Cohnella. Int. J. Syst. Evol. Microbiol. 2008, 58, 1855–1859. [Google Scholar]
- Flores-Félix, J.D.; Carro, L.; Velázquez, E.; Valverde, Á.; Cerda-Castillo, E.; García-Fraile, P.; Rivas, R. Phyllobacterium endophyticum sp. nov., isolated from nodules of Phaseolus vulgaris. Int. J. Syst. Evol. Microbiol. 2013, 63, 821–826. [Google Scholar]
- Mansfield, S.D.; Mooney, C.; Saddler, J.N. Substrate and enzyme characteristics that limit cellulose hydrolysis. Biotechnol. Prog. 1999, 15, 804–816. [Google Scholar] [CrossRef]
- Sharma, B.; Sarkar, A.; Singh, P.; Singh, R.P. Agricultural utilization of biosolids: A review on potential effects on soil and plant grown. Waste Manag. 2017, 64, 117–132. [Google Scholar] [CrossRef]
- Menéndez, E.; García-Fraile, P.; Rivas, R. Biotechnological applications of bacterial cellulases. AIMS Bioeng. 2015, 2, 163–182. [Google Scholar] [CrossRef]
- Collin, T.; Gerday, C.; Feller, G. Xylanase, xylanase families and extremophilic xylanase. FEMS Microbiol. Rev. 2005, 29, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Yadav, P.K.; Yadav, S.; Yadav, K.D.S. α-L-Rhamnosidase: A review. Process Biochem. 2010, 45, 1226–1235. [Google Scholar] [CrossRef]
- Xavier, J.R.; Ramana, K.V.; Sharma, R.K. β-galactosidase: Biotechnological applications in food processing. J. Food Biochem. 2018, 42, e12564. [Google Scholar] [CrossRef]
- Sieber, S.A.; Marahiel, M.A. Learning from nature’s drug factories: Nonribosomal synthesis of macrocyclic peptides. J. Bacteriol. 2003, 185, 7036–7043. [Google Scholar] [CrossRef]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Felnagle, E.A.; Jackson, E.E.; Chan, Y.A.; Podevels, A.M.; Berti, A.D.; McMahon, M.D.; Thomas, M.G. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 2008, 5, 191–211. [Google Scholar] [CrossRef]
- Agrawal, S.; Acharya, D.; Adholeya, A.; Barrow, C.J.; Deshmukh, S.K. Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front. Pharm. 2017, 8, 828. [Google Scholar] [CrossRef]
- Gross, F.; Luniak, N.; Perlova, O.; Gaitatzis, N.; Jenke-Kodama, H.; Gerth, K.; Müller, R. Bacterial type III polyketide synthases: Phylogenetic analysis and potential for the production of novel secondary metabolites by heterologous expression in pseudomonads. Arch. Microbiol. 2006, 185, 28–38. [Google Scholar] [CrossRef]
- Katsuyama, Y.; Ohnishi, Y. Type III polyketide synthases in microorganisms. Methods Enzymol. 2012, 515, 359–377. [Google Scholar]
- Cardoso, L.A.; Karp, S.G.; Vendruscolo, F.; Kanno, K.Y.; Zoz, L.I.; Carvalho, J.C. Biotechnological production of carotenoids and their applications in food and pharmaceutical products. Carotenoids 2017, 125. [Google Scholar]
- Gateau, H.; Solymosi, K.; Marchand, J.; Schoefs, B. Carotenoids of microalgae used in food industry and medicine. Mini Rev. Med. Chem. 2017, 17, 1140–1172. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Substrate | Enzyme Assayed |
|---|---|
| PNP-α-L-arabinopyranoside | α-L-arabinosidase |
| PNP-β-L-arabinopyranoside | β-L-arabinosidase |
| PNP-β-D-cellobioside | Cellobiohydrolase |
| PNP-phosphate (at pH 5.0) | Phosphatase |
| PNP-phosphate (at pH 7.0) | Phosphatase |
| PNP-phosphate (at pH 8.5) | Phosphatase |
| PNP-bis-phosphate (at pH 7) | Bisphosphatase |
| PNP-bisphosphate (at pH 8.5) | Bisphosphatase |
| PNP-α-L-fucopyranoside | α-L-fucosidase |
| PNP-β-D-fucopyranoside | β-glucosidase |
| PNP-α-D-galactopyranoside | α-galactosidase |
| PNP-β-D-galactopyranoside | β-galactosidase |
| PNP-β-D-galacturonide | β-galacturonidase |
| PNP-α-D-glucopyranoside | α-glucosidase |
| PNP-β-D-glucopyranoside | β-glucosidase |
| PNP-β-D-glucuronide | β-glucuronidase |
| PNP-β-D-lactopyranoside | β-lactosidase |
| PNP-α-D-mannopyranoside | α-mannosidase |
| PNP-β-D-mannopyranoside | β-mannosidase |
| PNP-α-L-rhamnopyranoside | α-rhamnosidase |
| PNP-N-thio-β-D-glucopyranoside | β-glucosidase |
| PNP-α-D-xylopyranoside | α-xylosidase |
| PNP-β-L-xylopyranoside | β-xylosidase |
| Attributes | Value |
|---|---|
| Genome size (bp) | 5,221,427 |
| G+C content (%) | 62.3 |
| N50 value | 130,073 |
| L50 value | 12 |
| Number of contigs (with PEGs) | 88 |
| Number of subsystems | 335 |
| Number of coding sequences | 5,244 |
| Number of RNAs | 51 |
| Number of genes related to: | |
| Cofactors, vitamins, prosthetic groups, pigments | 151 |
| Cell wall and capsule | 45 |
| Virulence, disease, and defense | 55 |
| Potassium metabolism | 8 |
| Miscellaneous | 26 |
| Phages, prophages, transposable elements, plasmids | 16 |
| Membrane transport | 179 |
| Iron acquisition and metabolism | 10 |
| RNA metabolism | 41 |
| Nucleosides and nucleotides | 96 |
| Protein metabolism | 187 |
| Motility and chemotaxis | 16 |
| Regulation and cell signaling | 47 |
| Secondary metabolism | 6 |
| DNA metabolism | 94 |
| Fatty acids, lipids, and isoprenoids | 109 |
| Nitrogen metabolism | 39 |
| Dormancy and sporulation | 1 |
| Respiration | 109 |
| Stress response | 66 |
| Metabolism of aromatic compounds | 56 |
| Amino acids and derivatives | 367 |
| Sulfur metabolism | 42 |
| Phosphorus metabolism | 39 |
| Carbohydrates | 267 |
| CAZyme Family | Gene Count | Known CAZyme Activities |
|---|---|---|
| GHs | ||
| 1 | 5 | β-glucosidase (EC 3.2.1.21); β-galactosidase (EC 3.2.1.23); β-mannosidase (EC 3.2.1.25); β-glucuronidase (EC 3.2.1.31); β-xylosidase (EC 3.2.1.37); β-D-fucosidase (EC 3.2.1.38); exo-β-1,4-glucanase (EC 3.2.1.74); 6-phospho-β-galactosidase (EC 3.2.1.85); and others |
| 2 | 2 | β-galactosidase (EC 3.2.1.23); β-mannosidase (EC 3.2.1.25); β-glucuronidase (EC 3.2.1.31); α-L-arabinofuranosidase (EC 3.2.1.55); exo-β-glucosaminidase (EC 3.2.1.165); α-L-arabinopyranosidase (EC 3.2.1.-); β-galacturonidase (EC 3.2.1.-); β-xylosidase (EC 3.2.1.37) |
| 3 | 2 | β-glucosidase (EC 3.2.1.21); xylan 1,4-β-xylosidase (EC 3.2.1.37); β-glucosylceramidase (EC 3.2.1.45); β-N-acetylhexosaminidase (EC 3.2.1.52); α-L-arabinofuranosidase (EC 3.2.1.55); glucan 1,3-β-glucosidase (EC 3.2.1.58); glucan 1,4-β-glucosidase (EC 3.2.1.74); and others |
| 15 | 1 | glucoamylase (EC 3.2.1.3); glucodextranase (EC 3.2.1.70); α,α-trehalase (EC 3.2.1.28); dextran dextrinase (EC 2.4.1.2) |
| 16 | 3 | xyloglucan:xyloglucosyltransferase (EC 2.4.1.207); keratan-sulfate endo-1,4-β-galactosidase (EC 3.2.1.103); endo-1,3-β-glucanase (EC 3.2.1.39); endo-1,3(4)-β-glucanase (EC 3.2.1.6); licheninase (EC 3.2.1.73); and others |
| 23 | 3 | lysozyme type G (EC 3.2.1.17); peptidoglycan lyase (EC 4.2.2.1), also known in the literature as peptidoglycan lytic transglycosylase; chitinase (EC 3.2.1.14) |
| 25 | 1 | lysozyme (EC 3.2.1.17) |
| 31 | 3 | α-glucosidase (EC 3.2.1.20); α-galactosidase (EC 3.2.1.22); α-mannosidase (EC 3.2.1.24); α-1,3-glucosidase (EC 3.2.1.84); sucrase-isomaltase (EC 3.2.1.48); α-xylosidase (EC 3.2.1.177); α-glucan lyase (EC 4.2.2.13); and others |
| 63 | 1 | α-glucosidase (EC 3.2.1.106); α-1,3-glucosidase (EC 3.2.1.84); α-glucosidase (EC 3.2.1.20); mannosylglycerate α-mannosidase / mannosylglycerate hydrolase (EC 3.2.1.170) |
| 102 | 1 | peptidoglycan lytic transglycosylase (EC 3.2.1.-) |
| 103 | 3 | peptidoglycan lytic transglycosylase (EC 3.2.1.-) |
| 105 | 1 | unsaturated rhamnogalacturonyl hydrolase (EC 3.2.1.172); d-4,5-unsaturated β-glucuronyl hydrolase (EC 3.2.1.-); d-4,5-unsaturated α-galacturonidase (EC 3.2.1.-) |
| 108 | 1 | N-acetylmuramidase (EC 3.2.1.17) |
| 109 | 2 | α-N-acetylgalactosaminidase (EC 3.2.1.49) |
| 113 | 1 | β-mannanase (EC 3.2.1.78) |
| PLs | ||
| 9 | 1 | pectate lyase (EC 4.2.2.2); exopolygalacturonate lyase (EC 4.2.2.9); thiopeptidoglycan lyase (EC 4.2.2.-) |
| 20 | 1 | endo-β-1,4-glucuronan lyase (EC 4.2.2.14) |
| GTs | ||
| 1 | 1 | UDP-glucuronosyltransferase (EC 2.4.1.17); zeatin O-β-xylosyltransferase (EC 2.4.2.40); 2-hydroxyacylsphingosine 1-β-galactosyltransferase (EC 2.4.1.45); N-acylsphingosine galactosyltransferase (EC 2.4.1.47); and others |
| 2 | 11 | cellulose synthase (EC 2.4.1.12); chitin synthase (EC 2.4.1.16); dolichyl-phosphate β-D-mannosyltransferase (EC 2.4.1.83); dolichyl-phosphate β-glucosyltransferase (EC 2.4.1.117); N-acetylglucosaminyltransferase (EC 2.4.1.-); and others |
| 4 | 19 | sucrose synthase (EC 2.4.1.13); sucrose-phosphate synthase (EC 2.4.1.14); α-glucosyltransferase (EC 2.4.1.52); lipopolysaccharide N-acetylglucosaminyltransferase (EC 2.4.1.56); phosphatidylinositol α-mannosyltransferase (EC 2.4.1.57); and others |
| 19 | 1 | lipid-A-disaccharide synthase (EC 2.4.1.182) |
| 20 | 1 | α,α-trehalose-phosphate synthase [UDP-forming] (EC 2.4.1.15); glucosylglycerol-phosphate synthase (EC 2.4.1.213); trehalose-6-P phosphatase (EC 3.1.3.12); [retaining] GDP-valeniol: validamine 7-phosphate valeniolyltransferase (EC 2.-.-.-) |
| 27 | 1 | polypeptide α-N-acetylgalactosaminyltransferase (EC 2.4.1.41) |
| 28 | 2 | 1,2-diacylglycerol 3-β-galactosyltransferase (EC 2.4.1.46); 1,2-diacylglycerol 3-β-glucosyltransferase (EC 2.4.1.157); UDP-GlcNAc: Und-PP-MurAc-pentapeptide β-N-acetylglucosaminyltransferase (EC 2.4.1.227); digalactosyldiacylglycerol synthase (EC 2.4.1.241) |
| 30 | 1 | CMP-β-KDO: α-3-deoxy-D-manno-octulosonic-acid (KDO) transferase (EC 2.4.99.-) |
| 51 | 6 | murein polymerase (EC 2.4.1.129) |
| 83 | 1 | undecaprenyl phosphate-α-L-Ara4N: 4-amino-4-deoxy-β-L-arabinosyltransferase (EC 2.4.2.43); dodecaprenyl phosphate-β-galacturonic acid: lipopolysaccharide core α-galacturonosyl transferase (EC 2.4.1.-) |
| 94 | 2 | GDP-Man: GlcA-β-1,2-Man-α-1,3-Glc-β-1,4-Glc-α-1-PP-undecaprenol β-1,4-mannosyltransferase (EC 2.4.1.251) |
| CEs | ||
| 1 | 4 | acetyl xylan esterase (EC 3.1.1.72); cinnamoyl esterase (EC 3.1.1.-); feruloyl esterase (EC 3.1.1.73); carboxylesterase (EC 3.1.1.1); S-formylglutathione hydrolase (EC 3.1.2.12); diacylglycerol O-acyltransferase (EC 2.3.1.20); trehalose 6-O-mycolyltransferase (EC 2.3.1.122) |
| 4 | 8 | acetyl xylan esterase (EC 3.1.1.72); chitin deacetylase (EC 3.5.1.41); chitooligosaccharide deacetylase (EC 3.5.1.-); peptidoglycan GlcNAc deacetylase (EC 3.5.1.-); peptidoglycan N-acetylmuramic acid deacetylase (EC 3.5.1.-) |
| 9 | 2 | N-acetylglucosamine 6-phosphate deacetylase (EC 3.5.1.25); N-acetylglucosamine 6-phosphate deacetylase (EC 3.5.1.80) |
| 10 | 6 | arylesterase (EC 3.1.1.-); carboxyl esterase (EC 3.1.1.3); acetylcholinesterase (EC 3.1.1.7); cholinesterase (EC 3.1.1.8); sterol esterase (EC 3.1.1.13); brefeldin A esterase (EC 3.1.1.-) |
| 11 | 1 | UDP-3-0-acyl N-acetylglucosamine deacetylase (EC 3.5.1.-) |
| 14 | 1 | N-acetyl-1-D-myo-inosityl-2-amino-2-deoxy-α-D-glucopyranoside deacetylase (EC 3.5.1.89); diacetylchitobiose deacetylase (EC 3.5.1.-); mycothiol S-conjugate amidase (EC 3.5.1.-) |
| AAs | ||
| 3 | 5 | cellobiose dehydrogenase (EC 1.1.99.18); glucose 1-oxidase (EC 1.1.3.4); aryl alcohol oxidase (EC 1.1.3.7); alcohol oxidase (EC 1.1.3.13); pyranose oxidase (EC 1.1.3.10) |
| 4 | 6 | vanillyl-alcohol oxidase (EC 1.1.3.38) |
| 6 | 1 | 1,4-benzoquinone reductase (EC 1.6.5.6) |
| 7 | 2 | glucooligosaccharide oxidase (EC 1.1.3.-); chitooligosaccharide oxidase (EC 1.1.3.-) |
| 12 | 1 | pyrroloquinoline quinone-dependent oxidoreductase |
| Fatty Acid | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| C16:0 | 9.2 | 4.8 | 6.6 | 7.6 |
| C17:0 | tr | 7.5 | ||
| C18:0 | 6.1 | 2.0 | 3.1 | 5.5 |
| C17:0 cyclo | 3.8 | 3.5 | 2.1 | tr |
| 19:0 cyclo ω8c | 24.3 | 57.7 | 11.8 | 27.9 |
| C20:2 ω6,9c | 1.1 | tr | 1.1 | |
| 11-Methyl C18:1 ω7c | 4.2 | 1.5 | tr | 1.5 |
| C18:0 3-OH | 1.7 | 2.1 | 1.1 | 1.7 |
| Summed feature 2 | 4.5 | 5.0 | 1.4 | 3.2 |
| Summed feature 3 | 4.5 | 1.1 | 5.2 | 1.1 |
| Summed feature 8 | 39.3 | 18.5 | 64.8 | 38.5 |
| Characteristic | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Isolation source | Plant | Soil | Air | Thermal aquifer |
| Colony color | White | Faint yellow | Light pink | Light pink |
| Motility | + | + | − | − |
| Oxidase | + | + | + | − |
| Nitrate reduction | + | + | − | + |
| Hydrolysis of: | ||||
| Gelatin | + | − | − | + |
| Aesculin | + | − | − | − |
| Assimilation of: | ||||
| D-Glucose | + | + | − | − |
| L-Arabinose | + | + | − | + |
| Production of: | ||||
| β-glucoronidase | − | − | + | − |
| Esterase lipase (c8) | + | w | + | + |
| Trypsin | + | − | + | w |
| pH range | 6–10 | 6–10 | 7–10 | 6–9 |
| Salinity range (%) | 0–1.5 | 0–1 | 0–2 | 0–1 |
| Temperature range (°C) | 12–37 | 15–37 | 10–35 | 25–45 |
| DNA G+C content (mol %) | 62.3 | 62.2 | 62.1 | 65.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Gómez, A.; Saati-Santamaría, Z.; Igual, J.M.; Rivas, R.; Mateos, P.F.; García-Fraile, P. Genome Insights into the Novel Species Microvirga brassicacearum, a Rapeseed Endophyte with Biotechnological Potential. Microorganisms 2019, 7, 354. https://doi.org/10.3390/microorganisms7090354
Jiménez-Gómez A, Saati-Santamaría Z, Igual JM, Rivas R, Mateos PF, García-Fraile P. Genome Insights into the Novel Species Microvirga brassicacearum, a Rapeseed Endophyte with Biotechnological Potential. Microorganisms. 2019; 7(9):354. https://doi.org/10.3390/microorganisms7090354
Chicago/Turabian StyleJiménez-Gómez, Alejandro, Zaki Saati-Santamaría, José M. Igual, Raúl Rivas, Pedro F. Mateos, and Paula García-Fraile. 2019. "Genome Insights into the Novel Species Microvirga brassicacearum, a Rapeseed Endophyte with Biotechnological Potential" Microorganisms 7, no. 9: 354. https://doi.org/10.3390/microorganisms7090354
APA StyleJiménez-Gómez, A., Saati-Santamaría, Z., Igual, J. M., Rivas, R., Mateos, P. F., & García-Fraile, P. (2019). Genome Insights into the Novel Species Microvirga brassicacearum, a Rapeseed Endophyte with Biotechnological Potential. Microorganisms, 7(9), 354. https://doi.org/10.3390/microorganisms7090354