Phylotypic Characterization of Mycobionts and Photobionts of Rock Tripe Lichen in East Antarctica



Abstract
:1. Introduction
2. Materials and Methods
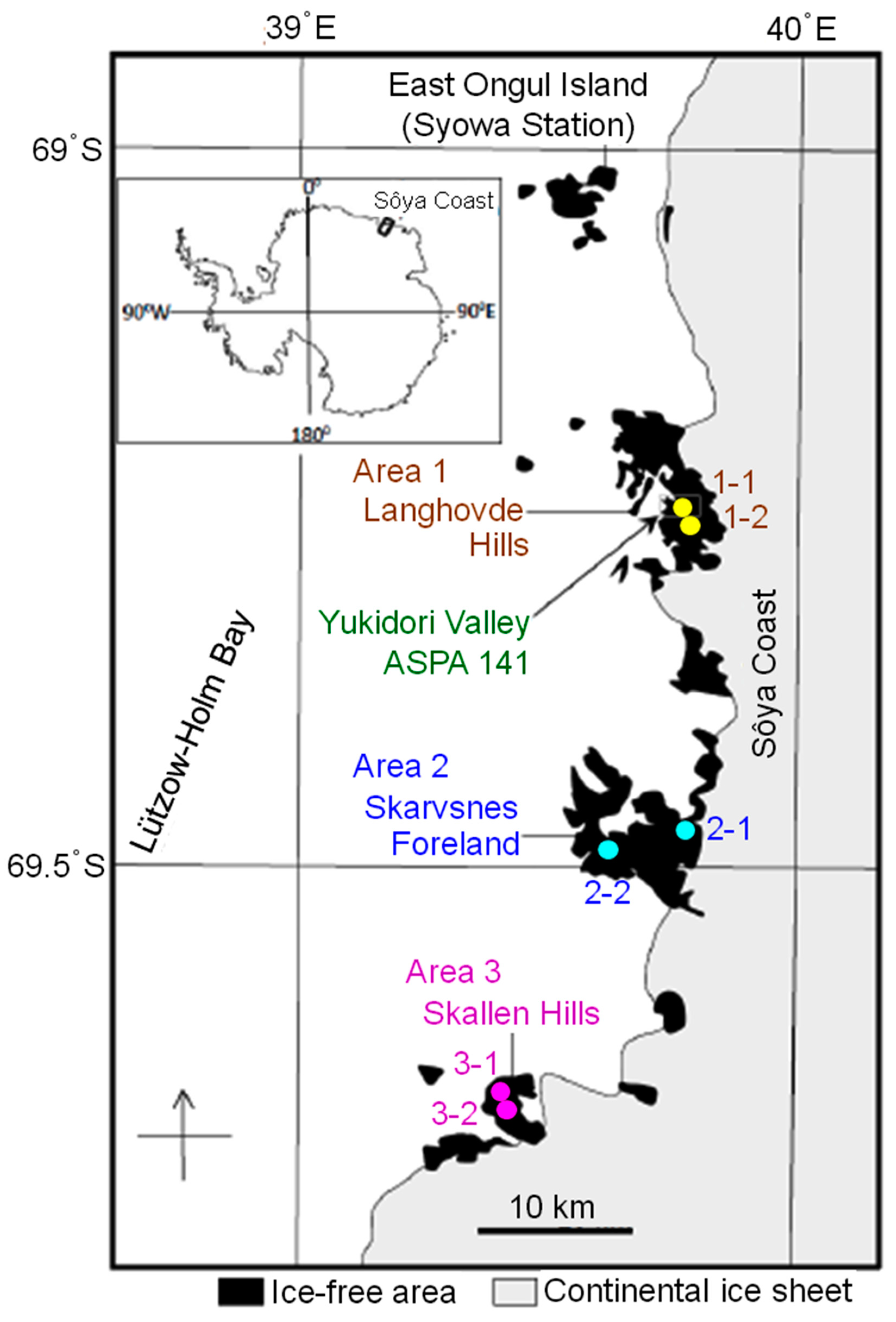
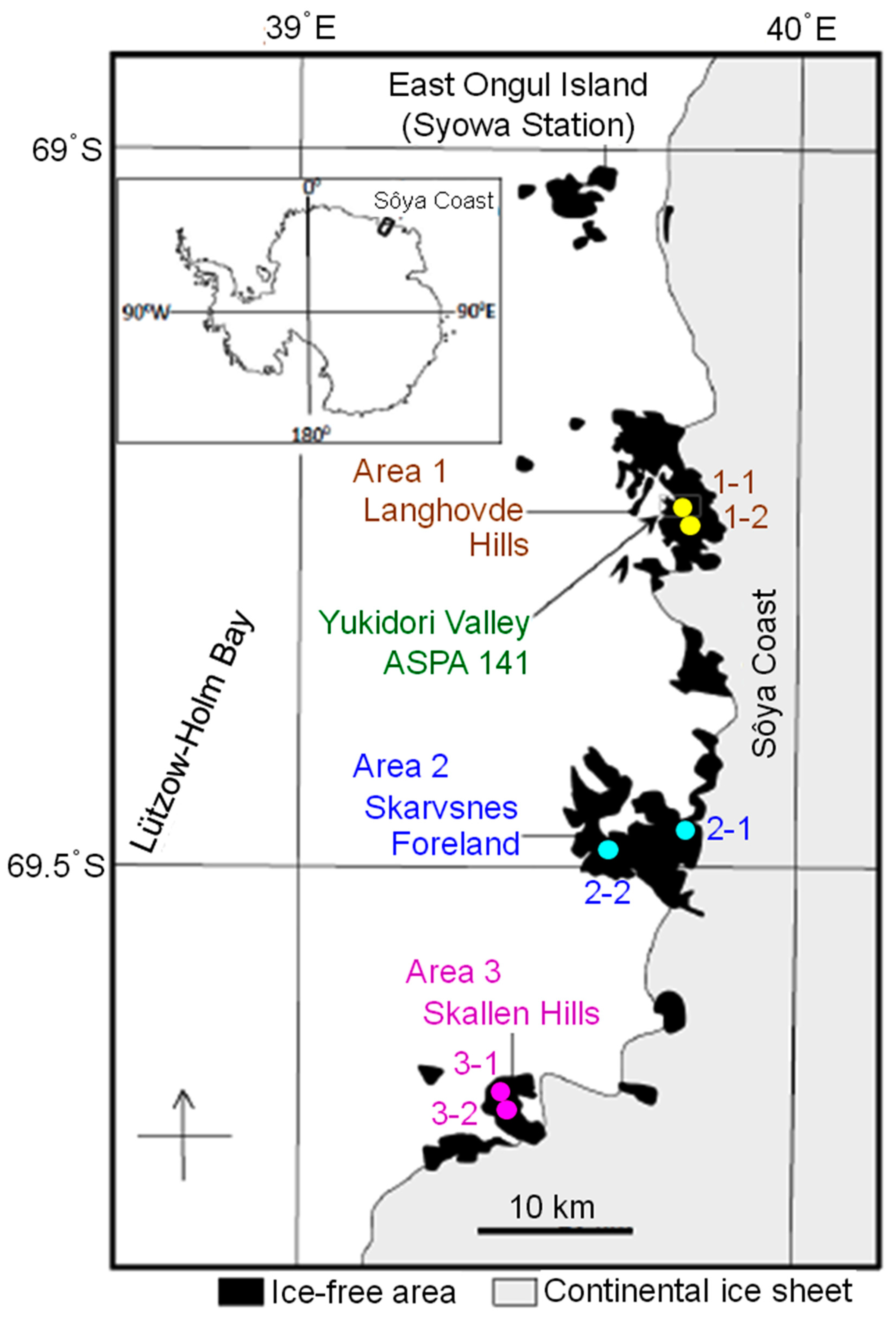
2.1. Collection of Rock Tripe Lichen Samples from Antarctica
2.2. DNA Extraction from the Lichen Thalli
2.3. Amplification, Cloning, and Sequencing of rRNA Gene-Related Sequences
2.3.1. PCR Amplification of rRNA Gene-Related Sequences
2.3.2. Cloning and Sequencing of rRNA Gene-Related Sequences
2.4. Massive Parallel Sequencing of V3-V4 Region of the 16S rRNA Genes From the Algae-Derived Chloroplasts, Cyanobacteria, and Other Bacteria
2.5. Data Analysis and Phylotype Determination
2.6. Diversity Indices and Phylogenetic Tree Analyses of the Phylotypes
3. Results and Discussion
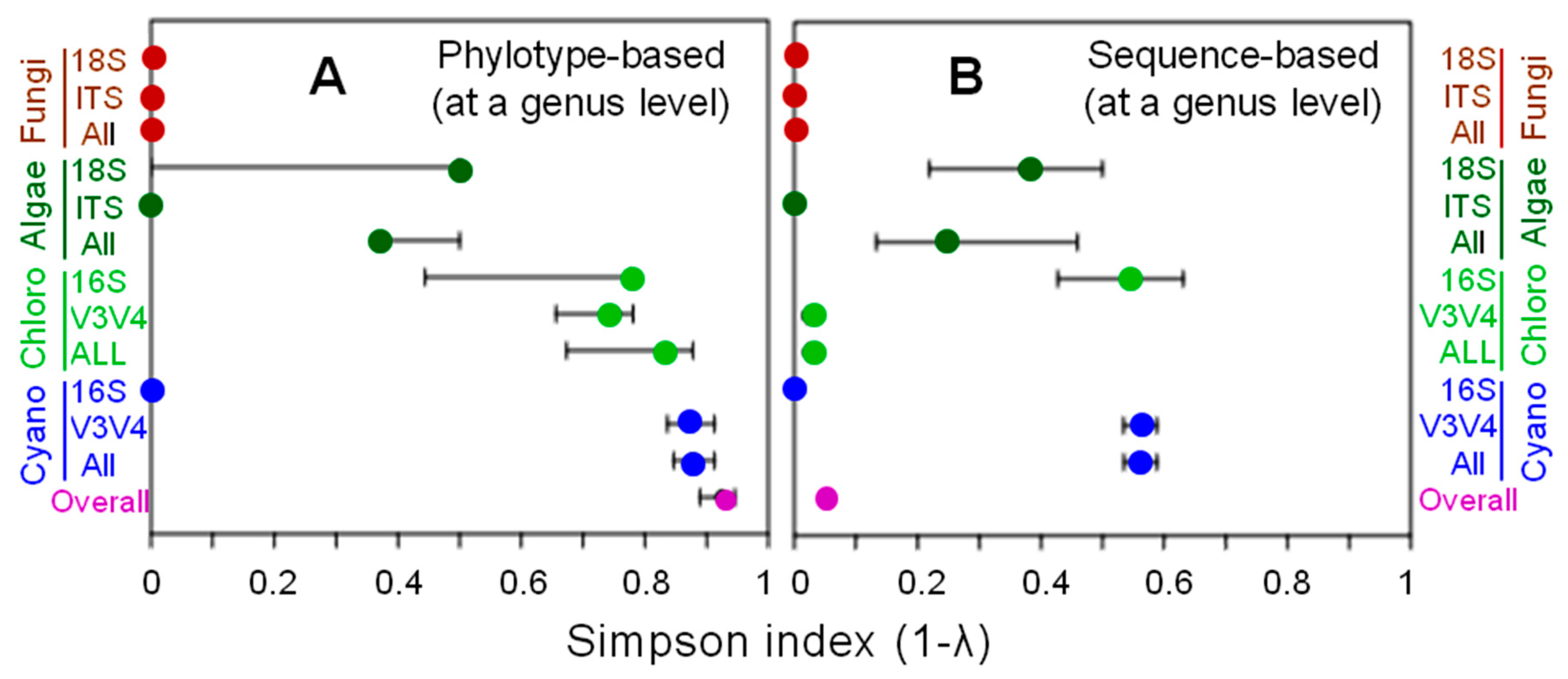
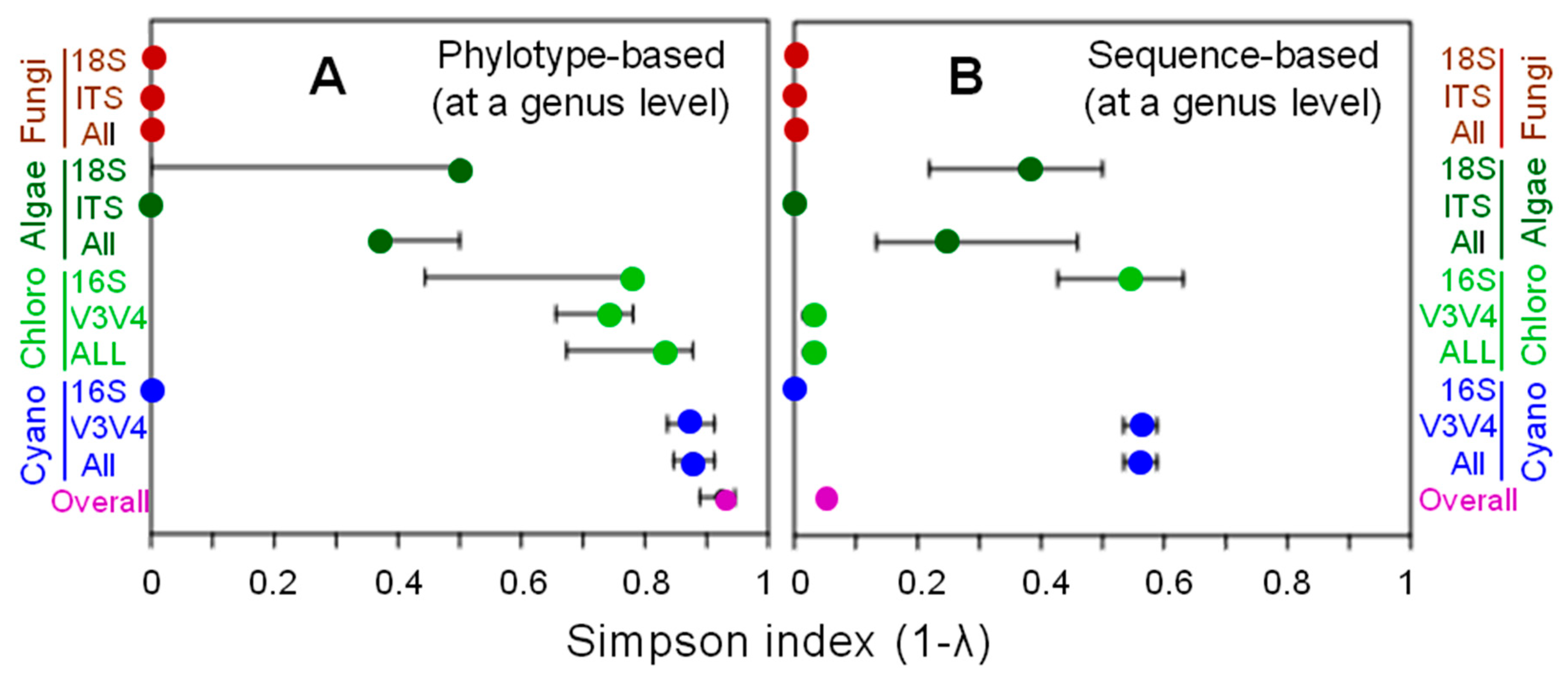
3.1. Phylotype Diversity
3.2. Phylotype Composition
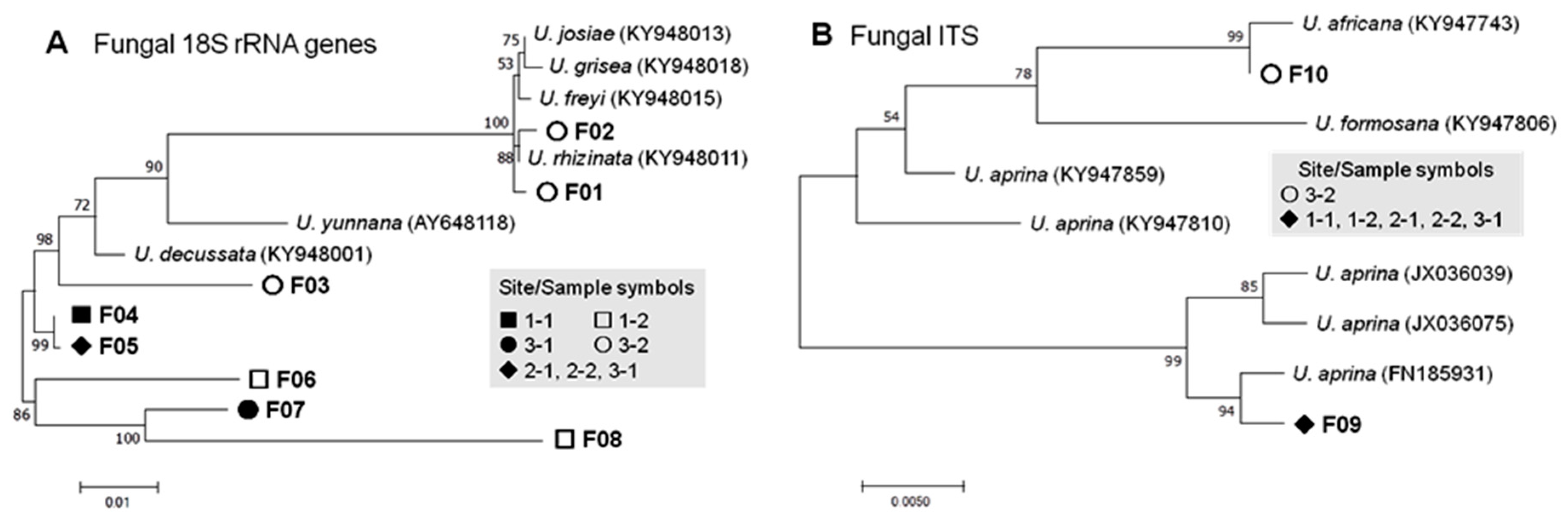
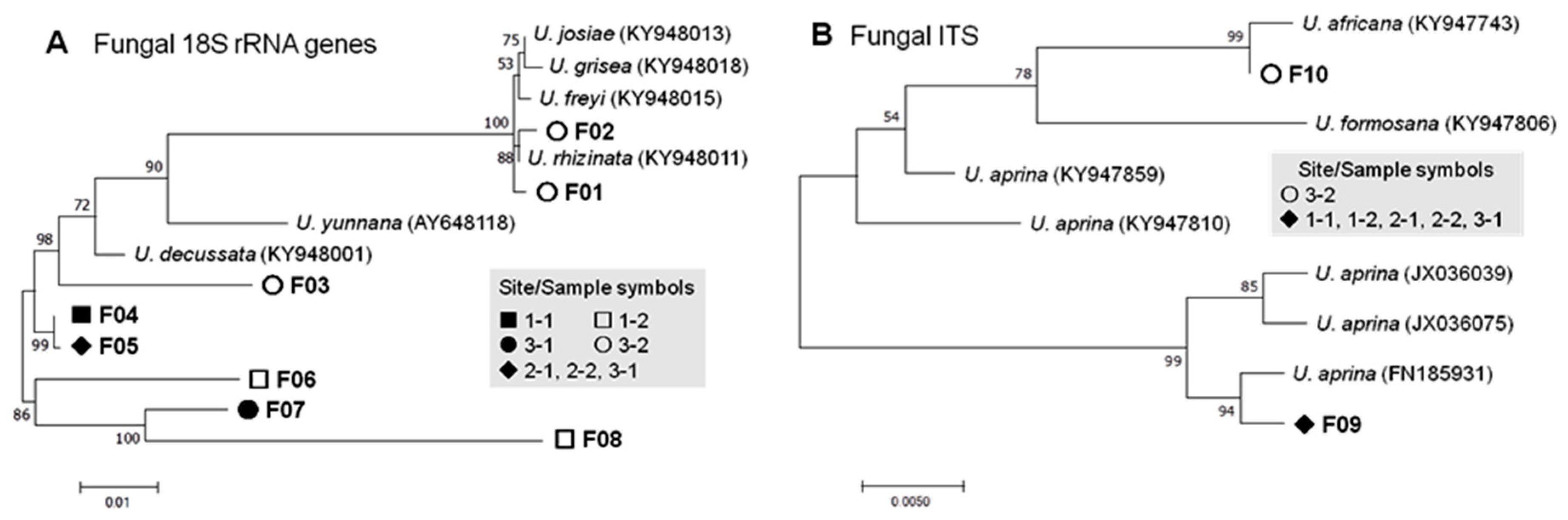
3.2.1. Fungal Phylotypes
3.2.2. Algal Phylotypes
3.2.3. Chloroplast Phylotypes
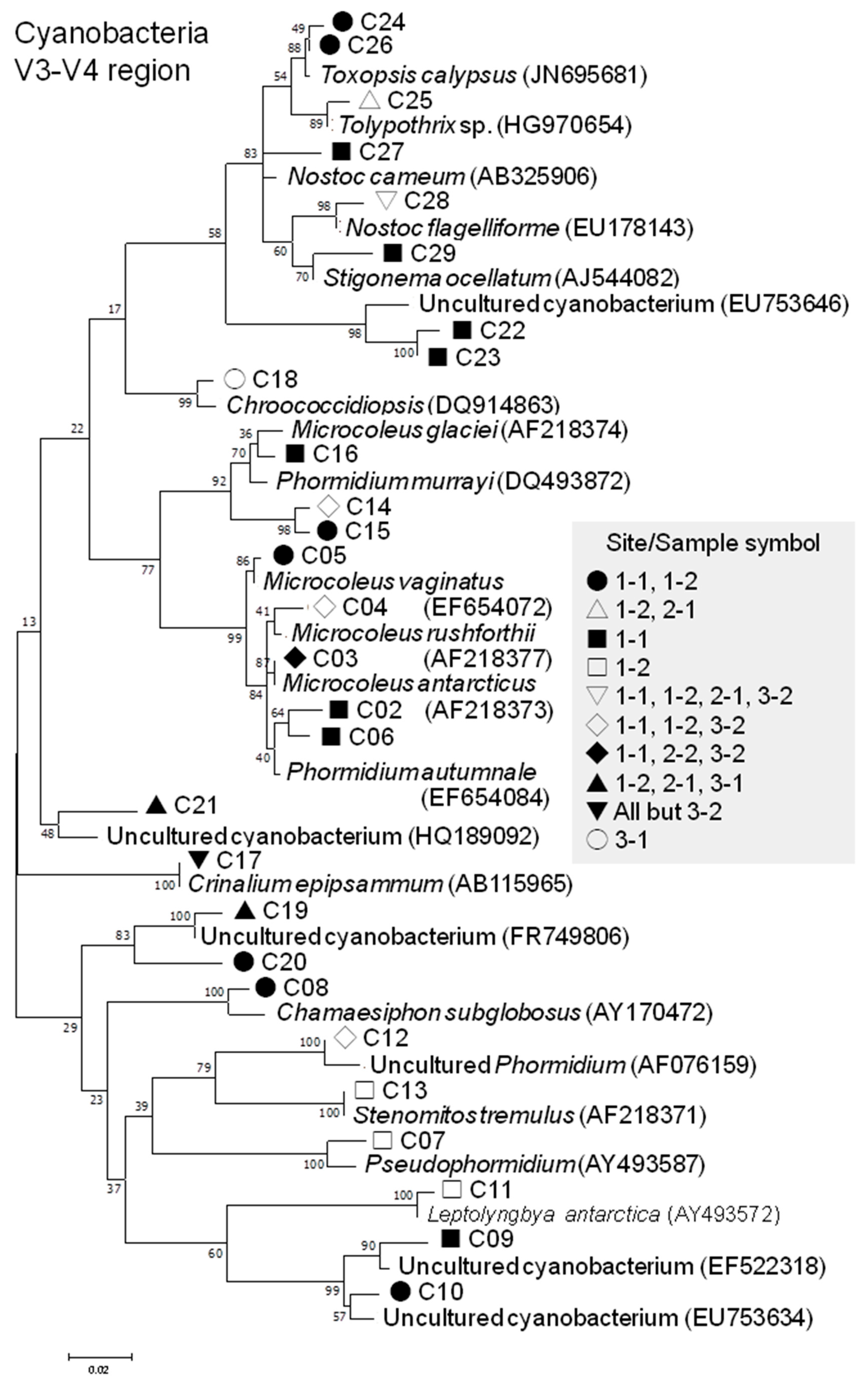
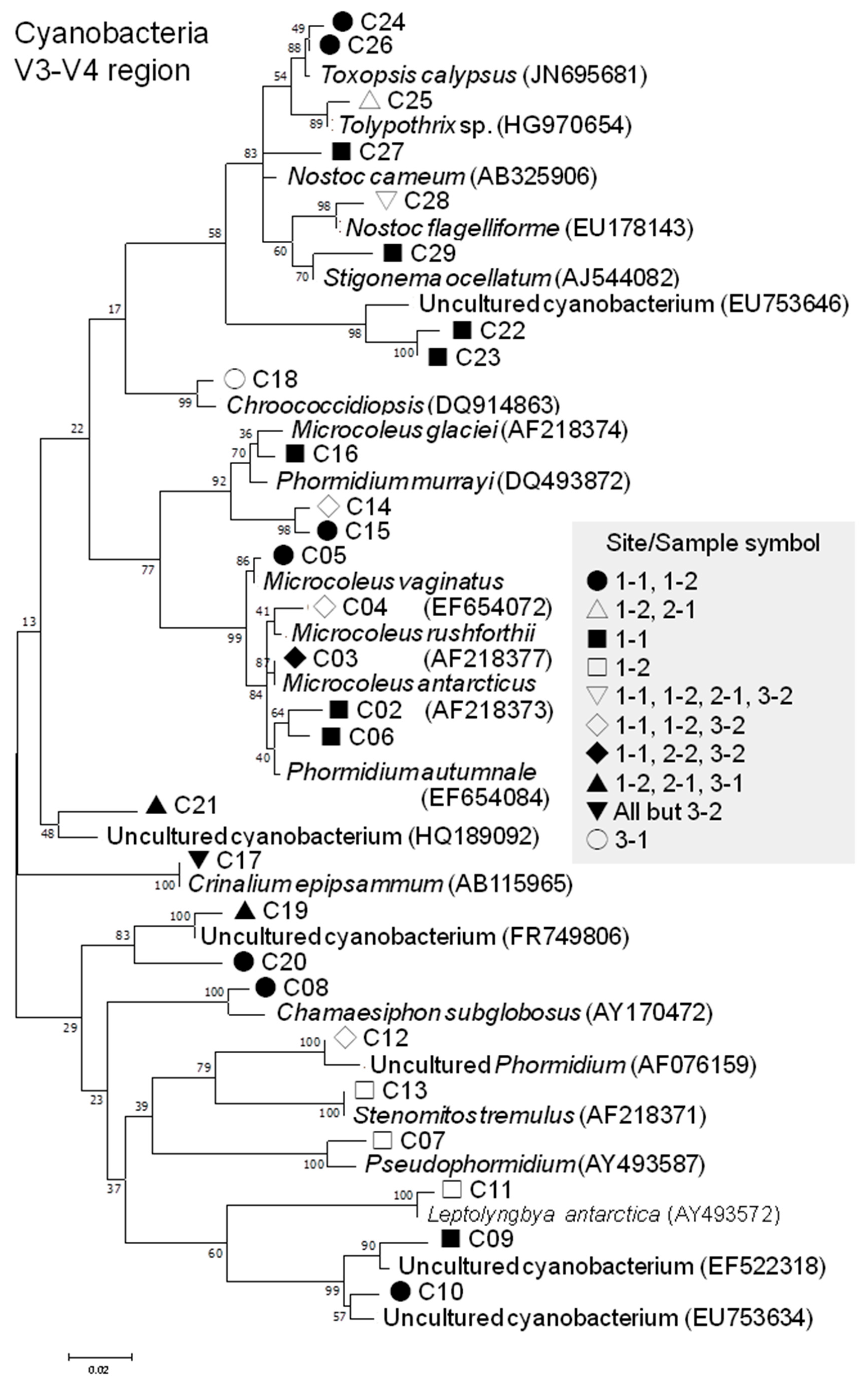
3.2.4. Cyanobacterial Phylotypes
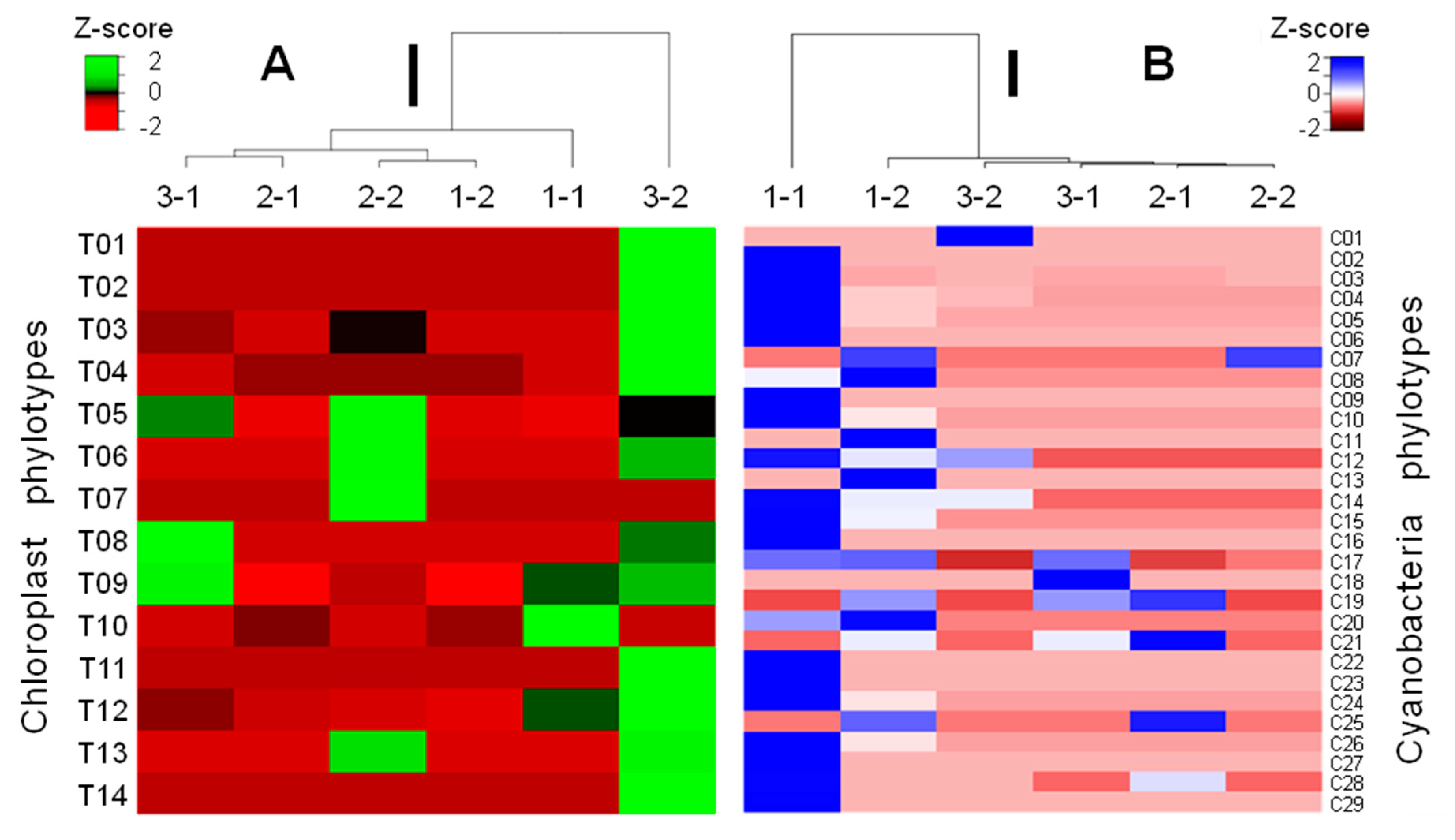
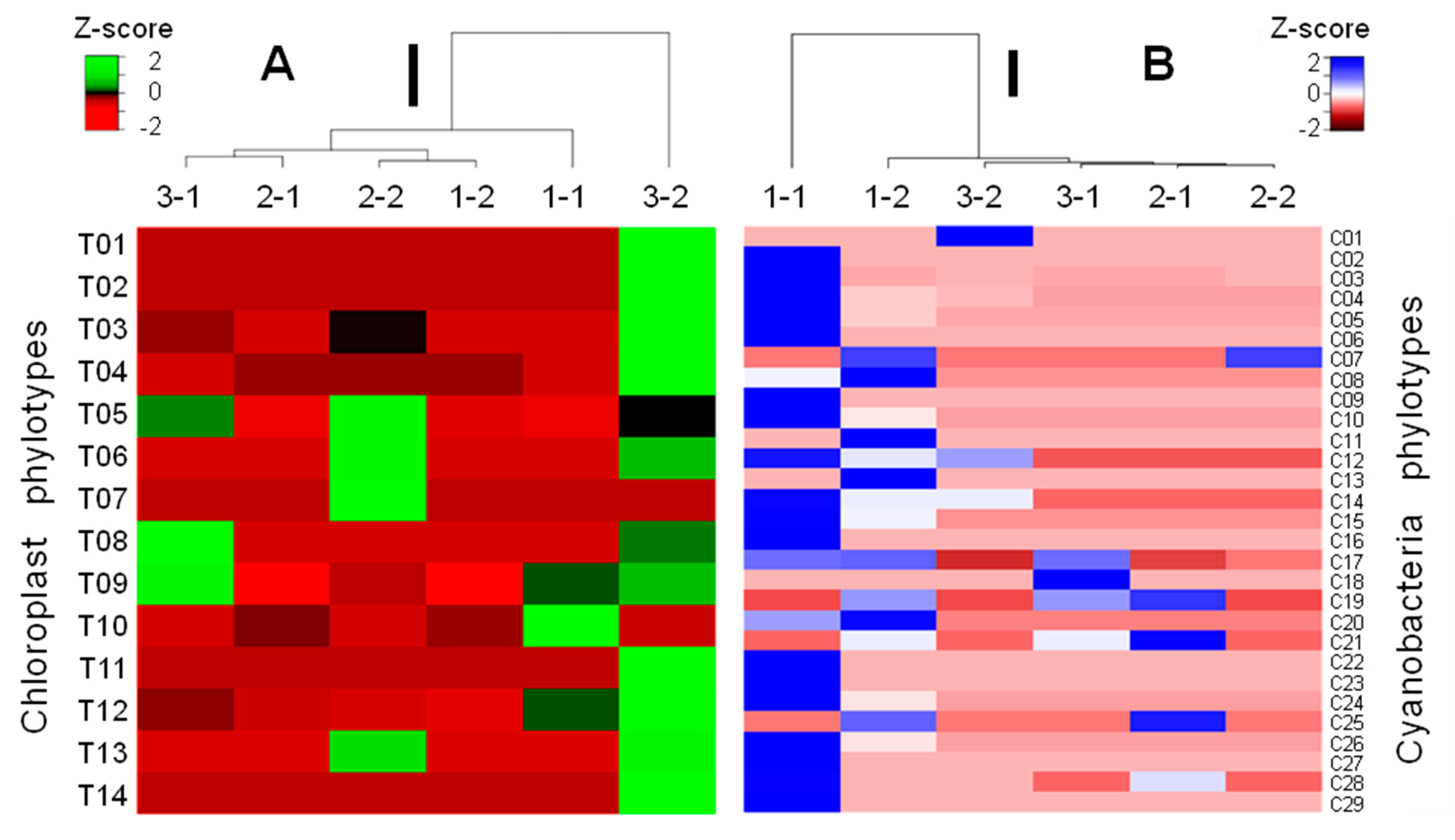
3.3. Phylotypic Profiles of the Studied Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Dictionary of the Fungi, 10th ed.; CABI: Wallingford, UK, 2008; p. 378. ISBN 978-0851998268. [Google Scholar]
- Friedl, T.; Büdel, B. Photobionts. In Lichen Biology, 2nd ed.; Nash, T.H., III, Ed.; Cambridge University Press: Cambridge, UK, 2012; pp. 9–26. [Google Scholar]
- Tschermak-Woess, E. The algal partner. In Handbook of Lichenology; Galun, M., Ed.; CRC Press: Boca Raton, FL, USA, 1988; Volume 1, pp. 39–92. ISBN 978-0849335815. [Google Scholar]
- Suryanarayanan, T.S.; Thirunavukkarasu, N. Endolichenic fungi: The lesser known fungal associates of lichens. Mycology 2017, 8, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Maduranga, K.; Attanayake, R.N.; Santhirasegaram, S.; Weerakoon, G.; Paranagama, P.A. Molecular phylogeny and bioprospecting of Endolichenic Fungi (ELF) inhabiting in the lichens collected from a mangrove ecosystem in Sri Lanka. PLoS ONE 2018, 13, e0200711. [Google Scholar] [CrossRef] [PubMed]
- Lawrey, J.D.; Diederich, P. Lichenicolous fungi: Interactions, evolution, and biodiversity. Bryologist 2003, 106, 80–120. [Google Scholar] [CrossRef]
- Spribille, T.; Tuovinen, V.; Resl1, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; Schneider, K.; Stabentheiner, E.; Toome-Heller, E.; Thor, G.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuovinen, V.; Ekman, S.; Thor, G.; Vanderpool, D.; Spribille, T.; Johannesson, H. Two basidiomycete fungi in the cortex of wolf lichens. Curr. Biol. 2019, 29, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Otte, J.; Schmitt, I.; Dal Grande, F. Comparing Sanger sequencing and high-throughput metabarcoding for inferring photobiont diversity in lichens. Sci. Rep. 2018, 8, 8624. [Google Scholar] [CrossRef] [Green Version]
- Farrar, J.F. Lichen as an ecosystem: Observation and experiment. In Lichenology: Progress and Problems; Brown, D.H., Hawksworth, D.L., Bailey, R.H., Eds.; Academic Press: London, UK, 1976; pp. 385–406. ISBN 978-0121367503. [Google Scholar]
- Parasyri, A.; Papazi, A.; Stamatis, N.; Zerveas, S.; Avramidou, E.V.; Doulis, A.G.; Pirintsos, S.; Kotzabasis, K. Lichen as micro-ecosystem: Extremophilic behavior with astrobiotechnological applications. Astrobiology 2018, 18, 1528–1542. [Google Scholar] [CrossRef]
- Richardson, D.H.S. War in the world of lichens: Parasitism and symbiosis as exemplified by lichens and lichenicolous fungi. Mycol. Res. 1999, 103, 641–650. [Google Scholar] [CrossRef]
- Wedin, M.; Maier, S.; Fernandez-Brime, S.; Cronholm, B.; Westberg, M.; Grube, M. Microbiome change by symbiotic invasion in lichens. Environ. Microbiol. 2016, 18, 1428–1439. [Google Scholar] [CrossRef]
- Haas, J.R.; Purvis, O.W. Lichen biogeochemistry. In Fungi in Biogeochemical Cycles; Gadd, G.M., Ed.; Cambridge University Press: Cambridge, UK, 2006; pp. 344–376. [Google Scholar]
- Lange, O.L.; Green, T.G.A. Lichens show that fungi can acclimate their respiration to seasonal changes in temperature. Oecologia 2005, 142, 11–19. [Google Scholar] [CrossRef]
- Gauslaa, Y.; Lie, M.; Solhaug, K.A.; Ohlson, M. Growth and ecophysiological acclimation of the foliose lichen Lobaria pulmonaria in forests with contrasting light climates. Oecologia 2006, 147, 406–416. [Google Scholar] [CrossRef]
- Pérez-Ortega, S.; Fernández-Mendoza, F.; Raggio, J.; Vivas, M.; Ascaso, C.; Sancho, L.G.; Printzen, C.; de Los Ríos, A. Extreme phenotypic variation in Cetraria aculeata (lichenized Ascomycota), adaptation or incidental modification? Ann. Bot. 2012, 109, 1133–1148. [Google Scholar] [CrossRef]
- Printzen, C.; Domaschke, S.; Fernández-Mendoza, F.; Pérez-Ortega, S. Biogeography and ecology of Cetraria aculeata, a widely distributed lichen with a bipolar distribution. MycoKeys 2013, 6, 33–53. [Google Scholar] [CrossRef]
- Piercey-Normore, M.D.; Depriest, P.T. Algal switching among lichen symbioses. Am. J. Bot. 2001, 88, 1490–1498. [Google Scholar] [CrossRef]
- Piercey-Normore, M.D. Selection of algal genotypes by three species of lichen fungi in the genus Cladonia. Can. J. Bot. 2004, 82, 947–961. [Google Scholar] [CrossRef]
- Blaha, J.; Baloch, E.; Grube, M. High photobiont diversity associated with the euryoecious lichen-forming ascomycete Lecanora rupicola (Lecanoraceae, Ascomycota). Biol. J. Linnean Soc. 2006, 88, 283–293. [Google Scholar] [CrossRef]
- Yahr, R.; Vilgalys, R.; Depriest, P.T. Geographic variation in algal partners of Cladonia subtenuis (Cladoniaceae) highlights the dynamic nature of a lichen symbiosis. New Phytol. 2006, 171, 847–860. [Google Scholar] [CrossRef]
- Castello, M.; Nimis, P. Diversity of lichens in Antarctica. In Antarctic Communities: Species, Structure and Survival; Valencia, J., Walton, D.W.H., Battaglia, B., Eds.; Cambridge University Press: Cambridge, UK, 2009; pp. 15–21. ISBN 978-0-521-11179-9. [Google Scholar]
- Green, T.G.A.; Lange, O.L. Photosynthesis in poikilohydric plants: A comparison of lichens and bryophytes. In Ecophysiology of Photosynthesis, Springer Study Edition, Volume 100; Schultze, E.-D., Caldwell, M.M., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; pp. 319–341. [Google Scholar]
- Sancho, L.G.; Pintado, A.; Green, T.G.A. Antarctic studies show lichens to be excellent biomonitors of climate change. Diversity 2019, 11, 42. [Google Scholar] [CrossRef]
- Ruprecht, U.; Brunauer, G.; Printzen, C. Genetic diversity of photobionts in Antarctic lecideoid lichens from an ecological view point. Lichenologist 2012, 44, 661–678. [Google Scholar] [CrossRef]
- Cardinale, M.; Puglia, A.M.; Grube, M. Molecular analysis of lichen-associated bacterial communities. FEMS Microbiol. Ecol. 2006, 57, 484–495. [Google Scholar] [CrossRef]
- Grube, M.; Cardinale, M.; de Castro, J.V., Jr.; Müller, H.; Berg, G. Species-specific structural and functional diversity of bacterial communities in lichen symbioses. ISME J. 2009, 3, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Bates, S.T.; Cropsey, G.W.; Caporaso, J.G.; Knight, R.; Fierer, N. Bacterial communities associated with the lichen symbiosis. Appl. Environ. Microbiol. 2011, 77, 1309–1314. [Google Scholar] [CrossRef]
- Cernava, T.; Berg, G.; Grube, M. High life expectancy of bacteria on lichens. Microb. Ecol. 2016, 72, 510–513. [Google Scholar] [CrossRef]
- Miller, D.N.; Bryant, J.E.; Madsen, E.L.; Ghiorse, W.C. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 1999, 65, 4715–4724. [Google Scholar]
- Medlin, L.; Elwood, H.J.; Stickel, S.; Sogin, M.L. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 1998, 30, 491–499. [Google Scholar] [CrossRef]
- Nakai, R.; Nakamura, K.; Jadoon, W.A.; Kashihara, K.; Naganuma, T. Genus-specific quantitative PCR of thraustochytrid protists. Mar. Ecol. Prog. Ser. 2013, 486, 1–12. [Google Scholar] [CrossRef]
- Delong, E.F. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. USA 1992, 89, 5685–5689. [Google Scholar] [CrossRef]
- Amann, R.I.; Binder, B.J.; Olson, R.J.; Chisholm, S.W.; Devereux, R.; Stahl, D.A. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 1990, 56, 1919–1925. [Google Scholar] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Yergeau, E.; Bell, T.H.; Champagne, J.; Maynard, C.; Tardif, S.; Tremblay, J.; Greer, C.W. Transplanting soil microbiomes leads to lasting effects on willow growth, but not on the rhizosphere microbiome. Front. Microbiol. 2015, 6, 1436. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Jumpponen, A. Soil fungal communities underneath willow canopies on a primary successional glacier forefront: rDNA sequence results can be affected by primer selection and chimeric data. Microb. Ecol. 2007, 53, 233–246. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.F.; Gao, Y.; Fu, L.M.; Li, W.Z. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Aronesty, E. Comparisons of sequencing utility programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Pajarillo, E.A.; Chae, J.P.; Kim, H.B.; Kim, I.H.; Kang, D.K. Barcoded pyrosequencing-based metagenomic analysis of the faecal microbiome of three purebred pig lines after cohabitation. Appl. Microbiol. Biotechnol. 2015, 99, 5647–5656. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, E.M.; Yi, M.H.; Lee, J.; Lee, S.; Hwang, Y.; Yong, D.; Sohn, W.M.; Yong, T.S. Intestinal fluke Metagonimus yokogawai infection increases probiotic Lactobacillus in mouse cecum. Exp. Parasitol. 2018, 193, 45–50. [Google Scholar] [CrossRef]
- Lee, B.; Moon, T.; Yoon, S.; Weissman, T. DUDE-Seq: Fast, flexible, and robust denoising for targeted amplicon sequencing. PLoS ONE 2017, 12, e0181463. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Unno, T. Bioinformatic suggestions on MiSeq-based microbial community analysis. J. Microbiol. Biotechnol. 2015, 25, 765–770. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiom. 2016, 2, 16004. [Google Scholar] [CrossRef]
- Mysara, M.; Vandamme, P.; Props, R.; Kerckhof, F.M.; Leys, N.; Boon, N.; Raes, J.; Monsieurs, P. Reconciliation between operational taxonomic units and species boundaries. FEMS Microbiol. Ecol. 2017, 93, fix029. [Google Scholar] [CrossRef]
- Wu, S.; Xiong, J.; Yu, Y. Taxonomic resolutions based on 18S rRNA genes: A case study of subclass Copepoda. PLoS ONE 2015, 10, e0131498. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS variability in the kingdom Fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, 193–201. [Google Scholar] [CrossRef]
- Lindeque, P.K.; Parry, H.E.; Harmer, R.A.; Somerfield, P.J.; Atkinson, A. Next generation sequencing reveals the hidden diversity of zooplankton assemblages. PLoS ONE 2013, 8, e81327. [Google Scholar] [CrossRef]
- Lemons, A.R.; Barnes, C.S.; Green, B.J. Comparative analysis of Sanger and Illumina Miseq sequencing for determining indoor fungal diversity. J. Allergy Clin. Immunol. 2017, 139, 86. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Krzewicka, B.; Smykla, J. The lichen genus Umbilicaria from the neighbourhood of Admiralty Bay (King George Island, maritime Antarctic), with a proposed new key to all Antarctic taxa. Polar Biol. 2004, 28, 15–25. [Google Scholar] [CrossRef]
- Krzewicka, B. Umbilicaria rhizinata comb. nov. (lichenized Ascomycota). Lichenologist 2010, 42, 491–493. [Google Scholar] [CrossRef]
- Beyer, L.; Bölter, M.; Seppelt, R.D. Nutrient and thermal regime, microbial biomass, and vegetation of Antarctic soils in the Windmill Islands region of East Antarctica (Wilkes Land). Arct. Antarct. Alp. Res. 2000, 32, 30–39. [Google Scholar] [CrossRef]
- Krzewicka, B.; García, M.A.; Johansen, S.D.; Sancho, L.G.; Martín, M.P. Morphological and nuclear ribosomal DNA data support distinguishing two new species of Umbilicaria (Umbilicariaceae, Ascomycota) from Europe. Lichenologist 2009, 41, 631–648. [Google Scholar] [CrossRef]
- Davydov, E.A.; Peršoh, D.; Rambold, G. Umbilicariaceae (lichenized Ascomycota)—Trait evolution and a new generic concept. Taxon 2017, 66, 1282–1303. [Google Scholar] [CrossRef]
- Nayaka, S.; Upreti, D.K. Schirmacher Oasis, East Antarctica, a lichenologically interesting region. Curr. Sci. 2005, 89, 1069–1071. [Google Scholar]
- Harańczyk, H.; Nowak, P.; Bacior, M.; Lisowska, M.; Marzec, M.; Florek, M.; Olech, M.A. Bound water freezing in Antarctic Umbilicaria aprina from Schirmacher Oasis. Antarct. Sci. 2012, 24, 342–352. [Google Scholar] [CrossRef]
- Khot, P.D.; Ko, D.L.; Fredricks, D.N. Sequencing and analysis of fungal rRNA operons for development of broad-range fungal PCR assays. Appl. Environ. Microbiol. 2009, 75, 1559–1565. [Google Scholar] [CrossRef]
- Darienko, T.; Gustavs, L.; Eggert, A.; Wolf, W.; Pröschold, T. Evaluating the species boundaries of green microalgae (Coccomyxa, Trebouxiophyceae, Chlorophyta) using integrative taxonomy and DNA barcoding with further implications for the species identification in environmental samples. PLoS ONE 2015, 10, e0127838. [Google Scholar] [CrossRef]
- Darienko, T.; Gustavs, L.; Mudimu, O.; Menendez, C.R.; Schumann, R.; Karsten, U.; Friedl, T.; Pröschold, T. Chloroidium, acommon terrestrial coccoid green alga previously assigned to Chlorella (Trebouxiophyceae, Chlorophyta). Eur. J. Phycol. 2010, 4, 79–95. [Google Scholar] [CrossRef]
- Turmel, M.; Otis, C.; Lemieux, C. Dynamic evolution of the chloroplast genome in the green algal casses Pedinophyceae and Trebouxiophyceae. Genome Biol. Evol. 2015, 7, 2062–2082. [Google Scholar] [CrossRef]
- Turmel, M.; Otis, C.; Lemieux, C. The chloroplast genomes of the green algae Pedinomonas minor, Parachlorella kessleri, and Oocystis solitaria reveal a shared ancestry between the Pedinomonadales and Chlorellales. Mol. Biol. Evol. 2009, 26, 2317–2331. [Google Scholar] [CrossRef]
- Guiry, M.D.; Guiry, G.M. AlgaeBase. World-Wide Electronic Publication, National University of Ireland, Galway. Available online: www.algaebase.org (accessed on 12 June 2019).
- Thüs, H.; Muggia, L.; Pérez-Ortega, S.; Favero-Longo, S.E.; Joneson, S.; O’Brien, H.; Nelsen, M.P.; Duque-Thüs, R.; Grube, M.; Friedl, T.; et al. Revisiting photobiont diversity in the lichen family Verrucariaceae (Ascomycota). Eur. J. Phycol. 2011, 46, 399–415. [Google Scholar] [CrossRef] [Green Version]
- Taton, A.; Grubisic, S.; Ertz, D.; Hodgson, D.A.; Piccardi, R.; Biondi, N.; Tredici, M.R.; Mainini, M.; Losi, D.; Marinelli, F.; et al. Polyphasic study of Antarctic cyanobacterial strains. J. Phycol. 2006, 42, 1257–1270. [Google Scholar] [CrossRef]
- Hirano, M. Freshwater algae from Yukidori Zawa, near Syowa Station, Antarctica. Mem. Natl. Inst. Polar Res. Spec. Issue 1979, 11, 1–25. [Google Scholar]
- Marshall, W.A.; Chalmers, M.O. Airborne dispersal of Antarctic terrestrial algae and cyanobacteria. Ecography 1997, 20, 585–594. [Google Scholar] [CrossRef]
- Rikkinen, J. Molecular studies on cyanobacterial diversity in lichen symbioses. MycoKeys 2013, 6, 3–32. [Google Scholar] [CrossRef] [Green Version]
- Casamatta, D.A.; Johansen, J.R.; Vis, M.L.; Broadwater, S.T. Molecular and morphological characterization of ten polar and near-polar strains within the Oscillatoriales (Cyanobacteria). J. Phycol. 2005, 41, 421–438. [Google Scholar] [CrossRef]
- Davydov, D. Diversity of the Cyanoprokaryota in polar deserts of Rijpfjorden east coast, North-East Land (Nordaustlandet) Island, Spitsbergen. Algol. Stud. Arch. 2013, 142, 29–44. [Google Scholar] [CrossRef]
- Zúñiga, C.; Leiva, D.; Carú, M.; Orlando, J. Substrates of Peltigera lichens as a potential source of cyanobionts. Microb. Ecol. 2017, 74, 561–569. [Google Scholar] [CrossRef]
- Bates, S.T.; Nash, T.H., III; Garcia-Pichel, F. Patterns of diversity for fungal assemblages of biological soil crusts from the southwestern United States. Mycologia 2012, 104, 353–361. [Google Scholar] [CrossRef]
- Lamprinou, V.; Skaraki, K.; Kotoulas, G.; Economou-Amilli, A.; Pantazidou, A. Toxopsis calypsus gen. nov., sp. nov. (Cyanobacteria, Nostocales) from cave ‘Francthi’, Peloponnese, Greece: A morphological and molecular evaluation. Int. J. Syst. Evol. Microbiol. 2012, 62, 2870–2877. [Google Scholar] [CrossRef]
- Tytgat, B.; Verleyen, E.; Sweetlove, M.; D’hondt, S.; Clercx, P.; Van Ranst, E.; Peeters, K.; Roberts, S.; Namsaraev, Z.; Wilmotte, A.; et al. Bacterial community composition in relation to bedrock type and macrobiota in soils from the Sør Rondane Mountains, East Antarctica. FEMS Microbiol. Ecol. 2016, 92, fiw126. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Area | Site/Sample | Longitude | Latitude | No. of Sampled Colonies |
|---|---|---|---|---|
| 1. Langhovde Hills | 1-1 | 69˚14′38.9” S | 39˚44′59.3” E | 1 |
| 1-2 | 69˚15′38.0” S | 39˚47′03.7” E | 1 | |
| 2. Skarvsnes Foreland | 2-1 | 69˚27′37.2” S | 39˚47′15.6” E | 1 |
| 2-2 | 69˚29′27.0” S | 39˚36′09.6” E | 1 | |
| 3. Skallen Hills | 3-1 | 69˚40′22.0” S | 39˚24′11.1” E | 2 |
| 3-2 | 69˚40′28.4” S | 39˚24′14.0” E | 1 |
| Target Sequence | Primer Designation | Forward/Reverse | -mer | 5′ → 3′ | Expected Product Size | Ref |
|---|---|---|---|---|---|---|
| 18S rRNA gene | EukF | F | 21 | AACCTGGTTGATCCTGCCAGT | Fungi, 2.0 kbp Algae, 1.8 kbp | [34] |
| EukR | R | 21 | TGATCCTTCTGCAGGTTCACC | |||
| Eukaryotic internal transcribed spacer (ITS) | ITS1F | F | 22 | CTTGGTCATTTAGAGGAAGTAA | 800 bp | [36] |
| ITS4R | R | 20 | TCCTCCGCTTATTGATATGC | [37] | ||
| 16S rRNA gene | 27F | F | 20 | AGAGTTTGATCCTGGCTCAG | 1.5 kbp | [34] |
| 1492R | R | 19 | GGTTACCTTGTTACGACTT | |||
| V3-V4 of 16S rRNA gene | 341F | F | 17 | CCTACGGGNGGCWGCAG | 460 bp | [38] |
| 806R | R | 21 | GACTACHVGGGTATCTAATCC | [39] |
| Class | Order | Family | Genus | Phylotype Number (%) Codes | Sequence Number (%) | ||
|---|---|---|---|---|---|---|---|
| Fungi | 18S | Lecanoromycetes | Umbilicariales | Umbilicariaceae | Umbilicaria | 8 (14.0%) F01–F08 | 21 (0.018%) |
| ITS | Lecanoromycetes | Umbilicariales | Umbilicariaceae | Umbilicaria | 2 (3.5%) F09, F10 | 6 (0.005%) | |
| Algae | 18S | Trebouxiophyceae | Trebouxiales | Coccomyxaceae | Coccomyxa | 1 (1.8%) A01 | 2 (0.002%) |
| Trebouxiaceae | Trebouxia | 1 (1.8%) A02 | 6 (0.005%) | ||||
| ITS | Trebouxiophyceae | Trebouxiales | Trebouxiaceae | Trebouxia | 2 (3.5%) A03, A04 | 6 (0.005%) | |
| Chloro-plasts | 16S | Trebouxiophyceae | Trebouxiales | Coccomyxaceae | Coccomyxa | 2 (3.5%) T01, T02 | 4 (0.003%) |
| Trebouxiaceae | Myrmecia | 1 (1.8%) T05 | 60 (0.051%) | ||||
| Microthamniales | Microthamniaceae | Microthamnion | 1 (1.8%) T04 | 13 (0.011%) | |||
| Chlorellales | Oocystaceae | Planctonema | 1 (1.8%) T06 | 3 (0.003%) | |||
| Chlorophyceae | Chlamydomonadales | incertae sedis | Ettlia | 1 (1.8%) T03 | 13 (0.011%) | ||
| V3-V4 | Trebouxiophyceae | Chlorellales | Chlorellaceae | Chloroidium | 1 (1.8%) T11 | 3 (0.003%) | |
| Oocystaceae | Neglectella | 1 (1.8%) T12 | 114,009 (97.496%) | ||||
| incertae sedis | Picochlorum | 1 (1.8%) T07 | 2 (0.002%) | ||||
| Prasiolales | Prasiolaceae | Edaphochlorella | 3 (5.3%) T08–T10 | 1216 (1.040%) | |||
| Trebouxiales | Coccomyxaceae | Coccomyxa | 2 (3.5%) T13, T14 | 83 (0.071%) | |||
| Cyano-bacteria | 16S | Cyanophyceae | Synechococcales | Leptolyngbyaceae | Leptolyngbya | 1 (1.8%) C01 | 1 (0.001%) |
| V3-V4 | Cyanophyceae | Oscillatoriales | Oscillatoriaceae | Phormidium | 3 (5.3%) C02, C06, C12 | 55 (0.047%) | |
| Microcoleaceae | Microcoleus | 5 (8.8%) C03–C05 C14, C15 | 953 (0.815%) | ||||
| Pseudophormidium | 1 (1.8%) C07 | 2 (0.002%) | |||||
| Coleofasciculaceae | Wilmottia | 1 (1.8%) C16 | 34 (0.029%) | ||||
| Gomontiellaceae | Crinalium | 1 (1.8%) C17 | 57 (0.049%) | ||||
| Synechococcales | Chamaesiphonaceae | Chamaesiphon | 1 (1.8%) C08 | 5 (0.004%) | |||
| Leptolyngbyaceae | Leptolyngbya | 1 (1.8%) C11 | 2 (0.002%) | ||||
| Stenomitos | 1 (1.8%) C13 | 2 (0.002%) | |||||
| Chroococcidiopsi-dales | Chroococcidiopsi-daceae | Chroococcidiopsis | 1 (1.8%) C18 | 4 (0.003%) | |||
| Nostocales | Nostocacea | Nostoc | 2 (3.5%) C27, C28 | 17 (0.015%) | |||
| Godleyaceae | Toxopsis | 2 (3.5%) C24, C26 | 204 (0.174%) | ||||
| Tolypothrichaceae | Tolypothrix | 1 (1.8%) C25 | 12 (0.010%) | ||||
| Stigonemataceae | Stigonema | 1 (1.8%) C29 | 31 (0.027%) | ||||
| Uncultured / unclassified | 7 (12.3%) C09, C10, C19–C23 | 111 (0.094%) | |||||
| Seq: | Code | Closest sequence | Area 1 Langhovde | Area 2 Skarvsnes | Area 3 Skallen | Sum | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Organism | Accession no. | Similarity (%) | 1-1 | 1-2 | 2-1 | 2-2 | 3-1 | 3-2 | ||||
| Fungal | 18S | F01 | Umbilicaria rhizinata | KY948011 | 99.6 | 0 | 0 | 0 | 0 | 0 | 2 | 2 |
| F02 | U. rhizinata | KY948011 | 99.7 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | ||
| F03 | U. rhizinata | KY948001 | 99.2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | ||
| F04 | U. decussata | KY948001 | 99.2 | 3 | 0 | 0 | 0 | 0 | 0 | 3 | ||
| F05 | U. decussata | KY948001 | 97.1 | 0 | 1 | 4 | 3 | 1 | 0 | 9 | ||
| F06 | U. decussata | KY948001 | 93.9 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | ||
| F07 | U. decussata | KY948001 | 95.1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | ||
| F08 | U. decussata | KY948001 | 91.0 | 0 | 1 | 0 | 0 | 1 | 0 | 2 | ||
| ITS | F09 | U. aprina | FN185931 | 99.5 | 1 | 1 | 1 | 1 | 1 | 0 | 5 | |
| F10 | U. africana | KY947743 | 98.7 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | ||
| Fungal total | 10 phylotypes | 4 | 4 | 5 | 4 | 4 | 6 | 27 | ||||
| Algal | 18S | A01 | Coccomyxa viridis Trebouxiophyceae | HG973007 | 99.7 | 0 | 0 | 0 | 0 | 0 | 2 | 2 |
| A02 | Trebouxia sp. SAG 2463 | KM020032 | 99.7 | 0 | 0 | 0 | 0 | 0 | 6 | 6 | ||
| ITS | A03 | Trebouxia sp. URa2 | JN204815 | 100 | 1 | 1 | 1 | 1 | 0 | 0 | 4 | |
| A04 | Trebouxia sp. FP-2018 | MH299127 | 100 | 0 | 0 | 0 | 0 | 1 | 1 | 2 | ||
| Algal total | 4 phylotypes | 1 | 1 | 1 | 1 | 1 | 9 | 14 | ||||
| Chloroplasts | 16S | T01 | Coccomyxa glaronensis Trebouxiophyceae | AM292034 | 92.8 | 0 | 0 | 0 | 0 | 0 | 3 | 3 |
| T02 | Coccomyxa sp. Trebouxiophyceae | MF805805 | 90.1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | ||
| T03 | Ettlia pseudoalveolaris Chlamydomonadales | KM462869 | 87.6 | 0 | 0 | 0 | 2 | 1 | 10 | 13 | ||
| T04 | Microthamnion kuetzingianumTrebouxiophyceae | KM462876 | 84.6 | 0 | 1 | 1 | 1 | 0 | 10 | 13 | ||
| T05 | Myrmecia israelensis Trebouxiophyceae | KM462861 | 88.3 | 0 | 1 | 0 | 35 | 14 | 10 | 60 | ||
| T06 | Planctonema lauterbornii Trebouxiophyceae | KM462880 | 88.0 | 0 | 0 | 0 | 2 | 0 | 1 | 3 | ||
| V3-V4 | T07 | Picochlorum eukaryotum Trebouxiophyceae | X76084 | 81.9 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | |
| T08 | Edaphochlorella mirabilis Trebouxiophyceae | X65100 | 94.8 | 0 | 0 | 0 | 0 | 3 | 1 | 4 | ||
| T09 | Edaphochlorella mirabilis Trebouxiophyceae | X65100 | 92.1 | 212 | 89 | 116 | 158 | 348 | 260 | 1183 | ||
| T10 | Edaphochlorella mirabilis Trebouxiophyceae | X65100 | 94.8 | 23 | 2 | 3 | 0 | 0 | 1 | 29 | ||
| T11 | Chloroidium saccharophilum Trebouxiophyceae | D11349 | 97.7 | 0 | 0 | 0 | 0 | 0 | 3 | 3 | ||
| T12 | Neglectella solitaria Trebouxiophyceae | FJ968739 | 92.8 | 21,375 | 5772 | 10,596 | 7924 | 14,065 | 54,277 | 114,009 | ||
| T13 | Coccomyxa subellipsoidea Trebouxiophyceae | HQ693844 | 91.6 | 0 | 0 | 0 | 2 | 0 | 3 | 5 | ||
| T14 | Coccomyxasimplex Trebouxiophyceae | AM292034 | 95.7 | 0 | 0 | 0 | 0 | 0 | 78 | 78 | ||
| Chloroplast total | 14 phylotypes | 21,610 | 5865 | 10,716 | 8126 | 14,431 | 54,658 | 115,406 | ||||
| Cyanobacterial | 16S | C01 | Leptolyngbya antarctica Synechococcales | AY493590 | 98.2 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| V3-V4 | C02 | Phormidium autumnale Oscillatoriophycideae | EF654084 | 93.2 | 32 | 0 | 0 | 0 | 0 | 0 | 32 | |
| C03 | Microcoleus antarcticus Oscillatoriophycideae | AF218373 | 99.5 | 181 | 0 | 0 | 1 | 0 | 1 | 183 | ||
| C04 | Microcoleus rushforthii Oscillatoriophycideae | AF218377 | 99.0 | 478 | 34 | 0 | 0 | 0 | 19 | 531 | ||
| C05 | Microcoleus vaginatus Oscillatoriophycideae | EF654072 | 99.7 | 203 | 16 | 0 | 0 | 0 | 0 | 219 | ||
| C06 | Phormidium autumnale Oscillatoriophycideae | EF654084 | 98.1 | 9 | 0 | 0 | 0 | 0 | 0 | 9 | ||
| C07 | Pseudophormidium Oscillatoriophycideae | AY493587 | 97.6 | 0 | 1 | 0 | 1 | 0 | 0 | 2 | ||
| C08 | Chamaesiphon subglobosusSynechococcales | AY170472 | 97.4 | 1 | 4 | 0 | 0 | 0 | 0 | 5 | ||
| C09 | Uncultured cyanobacterium (from Rocky Mountains) | EF522318 | 98.6 | 22 | 0 | 0 | 0 | 0 | 0 | 22 | ||
| C10 | Uncultured cyanobacterium (from Spanish stromatolites) | EU753634 | 98.6 | 8 | 1 | 0 | 0 | 0 | 0 | 9 | ||
| C11 | Leptolyngbya atarctica Synechococcales | AY493572 | 99.5 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | ||
| C12 | Phormidium sp. Oscillatoriophycideae | AF076159 | 98.6 | 7 | 3 | 0 | 0 | 0 | 4 | 14 | ||
| C13 | Stenomitos tremulus Synechococcales | AF218371 | 94.6 | 0 | 2 | 0 | 0 | 0 | 0 | 2 | ||
| C14 | Microcoleus glaciei Oscillatoriophycideae | AF218374 | 96.9 | 6 | 2 | 0 | 0 | 0 | 2 | 10 | ||
| C15 | Microcoleus glaciei Oscillatoriophycideae | AF218374 | 97.2 | 8 | 2 | 0 | 0 | 0 | 0 | 10 | ||
| C16 | Wilmottia murrayi Oscillatoriophycideae | DQ493872 | 98.8 | 34 | 0 | 0 | 0 | 0 | 0 | 34 | ||
| C17 | Crinalium epipsammum Oscillatoriophycideae | AB115965 | 98.6 | 17 | 18 | 1 | 4 | 17 | 0 | 57 | ||
| C18 | Chroococcidiopsis Chroococcidiopsidales | DQ914863 | 96.0 | 0 | 0 | 0 | 0 | 4 | 0 | 4 | ||
| C19 | Uncultured cyanobacterium (from Antarctic Peninsula) | FR749806 | 99.3 | 0 | 4 | 6 | 0 | 4 | 0 | 14 | ||
| C20 | Uncultured cyanobacterium (from Antarctic Peninsula) | FR749806 | 98.8 | 1 | 2 | 0 | 0 | 0 | 0 | 3 | ||
| C21 | Uncultured cyanobacterium (from Antarctic Dry Valleys) | HQ189092 | 95.7 | 0 | 2 | 6 | 0 | 2 | 0 | 10 | ||
| C22 | Uncultured cyanobacterium (from Spanish stromatolites) | EU753646 | 96.7 | 27 | 0 | 0 | 0 | 0 | 0 | 27 | ||
| C23 | Uncultured cyanobacterium (from Spanish stromatolites) | EU753646 | 97.2 | 26 | 0 | 0 | 0 | 0 | 0 | 26 | ||
| C24 | Toxopsis calypsus Nostocales | JN695681 | 96.7 | 91 | 11 | 0 | 0 | 0 | 0 | 102 | ||
| C25 | Tolypothrix sp. Nostocales | HG970654 | 98.6 | 0 | 5 | 7 | 0 | 0 | 0 | 12 | ||
| C26 | Toxopsis calypsus Nostocales | JN695681 | 98.6 | 76 | 26 | 0 | 0 | 0 | 0 | 102 | ||
| C27 | Nostoc carneum Nostocales | AB325906 | 98.1 | 4 | 0 | 0 | 0 | 0 | 0 | 4 | ||
| C28 | Nostoc flagelliforme Nostocales | EU178143 | 98.6 | 8 | 1 | 3 | 0 | 0 | 1 | 13 | ||
| C29 | Stigonema ocellatum Nostocales | AJ544082 | 97.9 | 31 | 0 | 0 | 0 | 0 | 0 | 31 | ||
| Cyanobacterial total | 29 phylotypes | 1270 | 136 | 23 | 6 | 27 | 28 | 1490 | ||||
| Overall total | 57 phylotypes | 22,885 | 6006 | 10,745 | 8137 | 14,463 | 54,701 | 11,6937 | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faluaburu, M.S.; Nakai, R.; Imura, S.; Naganuma, T. Phylotypic Characterization of Mycobionts and Photobionts of Rock Tripe Lichen in East Antarctica. Microorganisms 2019, 7, 203. https://doi.org/10.3390/microorganisms7070203
Faluaburu MS, Nakai R, Imura S, Naganuma T. Phylotypic Characterization of Mycobionts and Photobionts of Rock Tripe Lichen in East Antarctica. Microorganisms. 2019; 7(7):203. https://doi.org/10.3390/microorganisms7070203
Chicago/Turabian StyleFaluaburu, Merry Sailonga, Ryosuke Nakai, Satoshi Imura, and Takeshi Naganuma. 2019. "Phylotypic Characterization of Mycobionts and Photobionts of Rock Tripe Lichen in East Antarctica" Microorganisms 7, no. 7: 203. https://doi.org/10.3390/microorganisms7070203
APA StyleFaluaburu, M. S., Nakai, R., Imura, S., & Naganuma, T. (2019). Phylotypic Characterization of Mycobionts and Photobionts of Rock Tripe Lichen in East Antarctica. Microorganisms, 7(7), 203. https://doi.org/10.3390/microorganisms7070203