Elevation is Associated with Human Skin Microbiomes


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Volunteer Recruitment and Sample Collection
2.2. DNA Extraction and High-Throughput Sequencing
2.3. Bioinformatics Analysis
2.4. Statistical Analysis
2.5. Co-Occurrence Patterns Analysis
2.6. Community Assembly Processes Analysis
2.7. Function Prediction of Skin Microbiota
3. Results
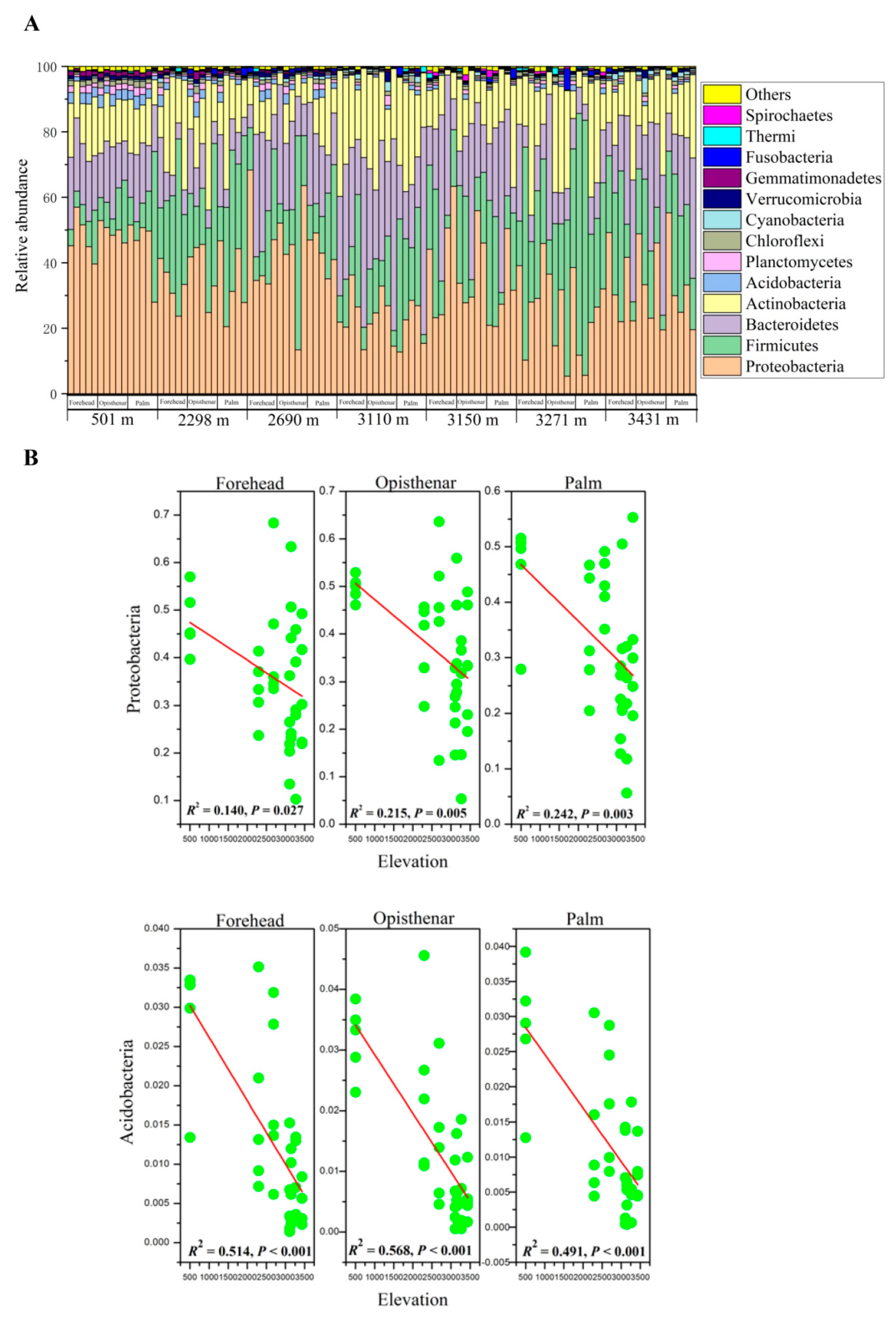
3.1. Overall Composition of Human Skin Microbiota
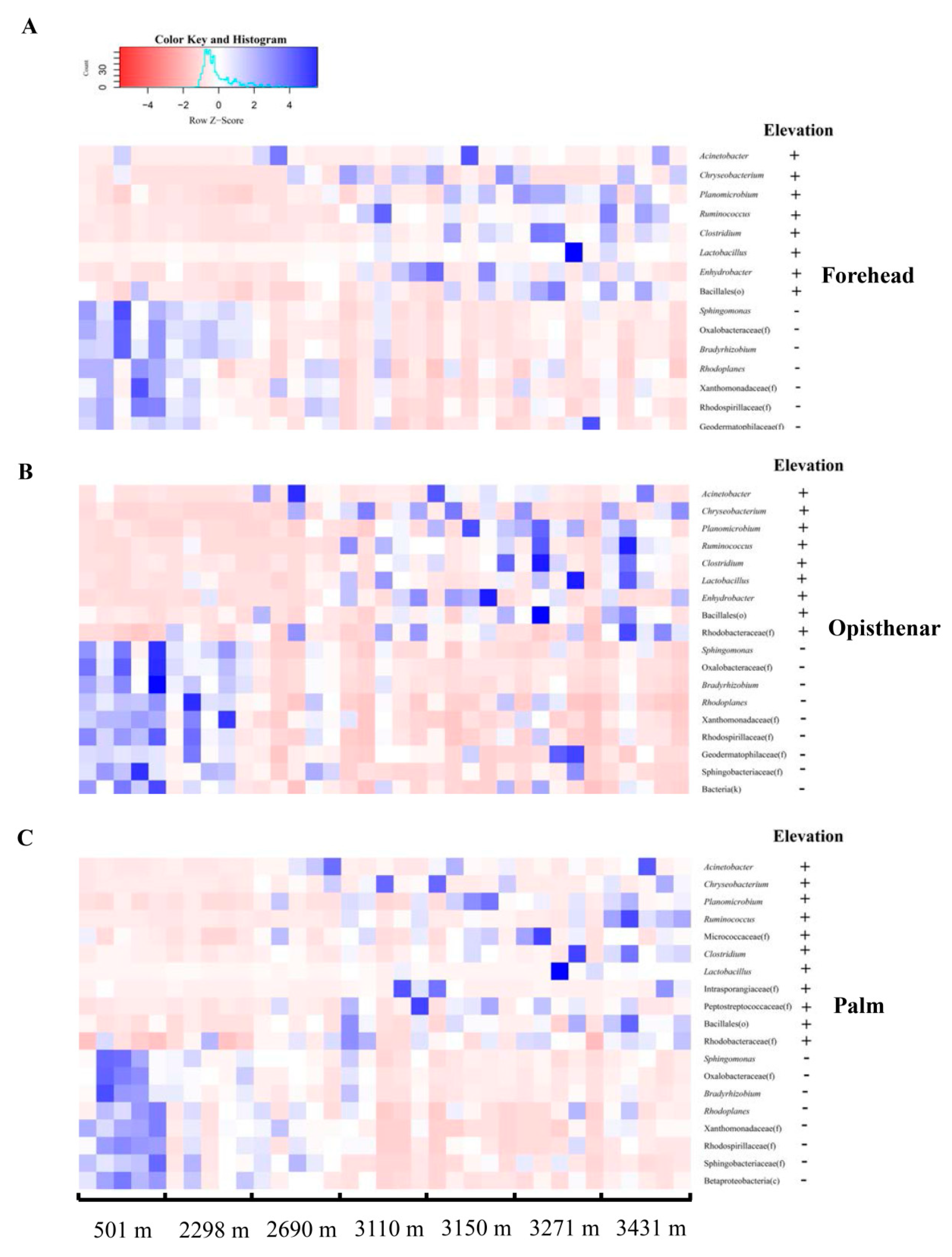
3.2. Elevation-Sensitive and Elevation-Tolerant Microbes
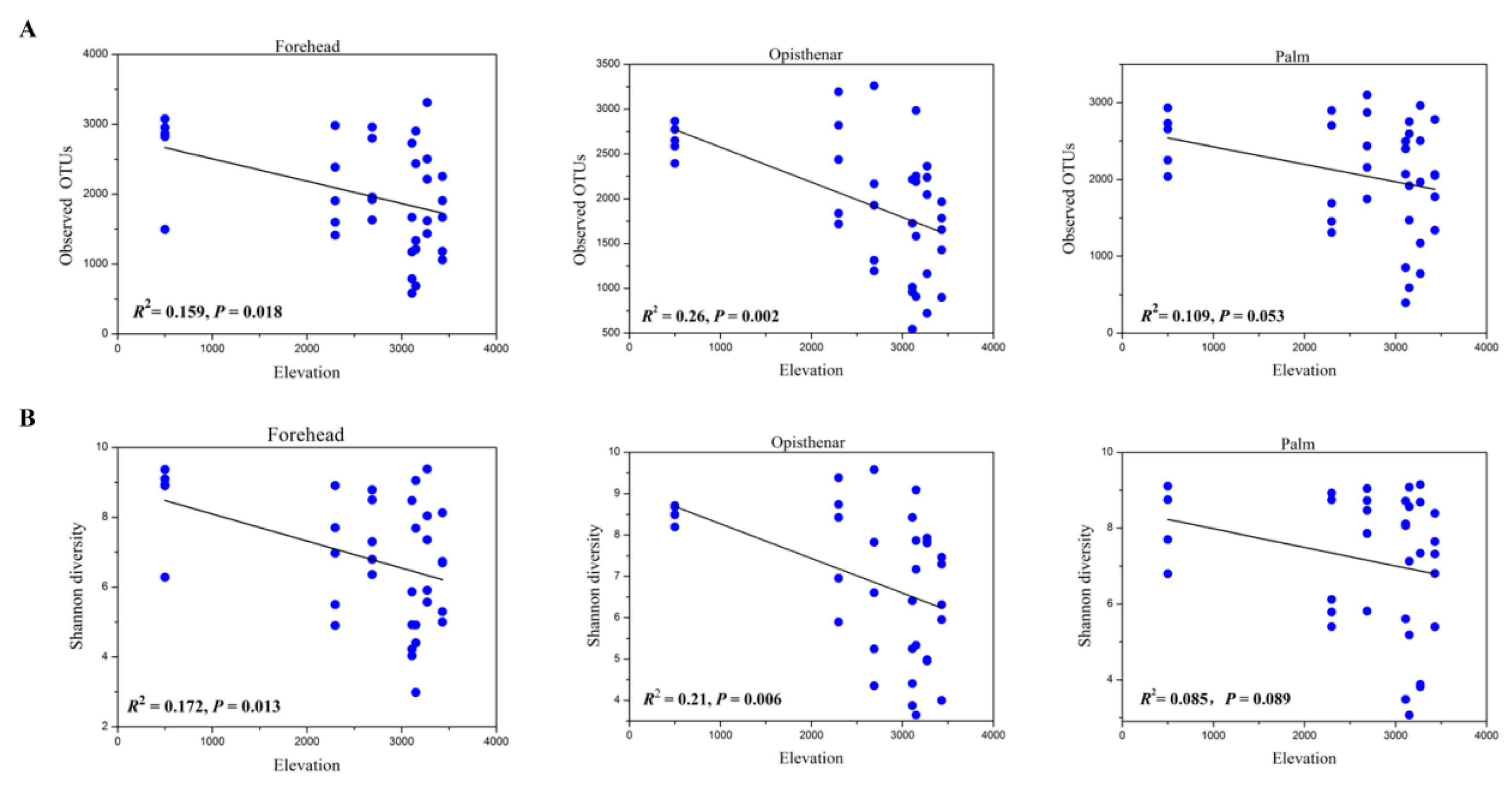
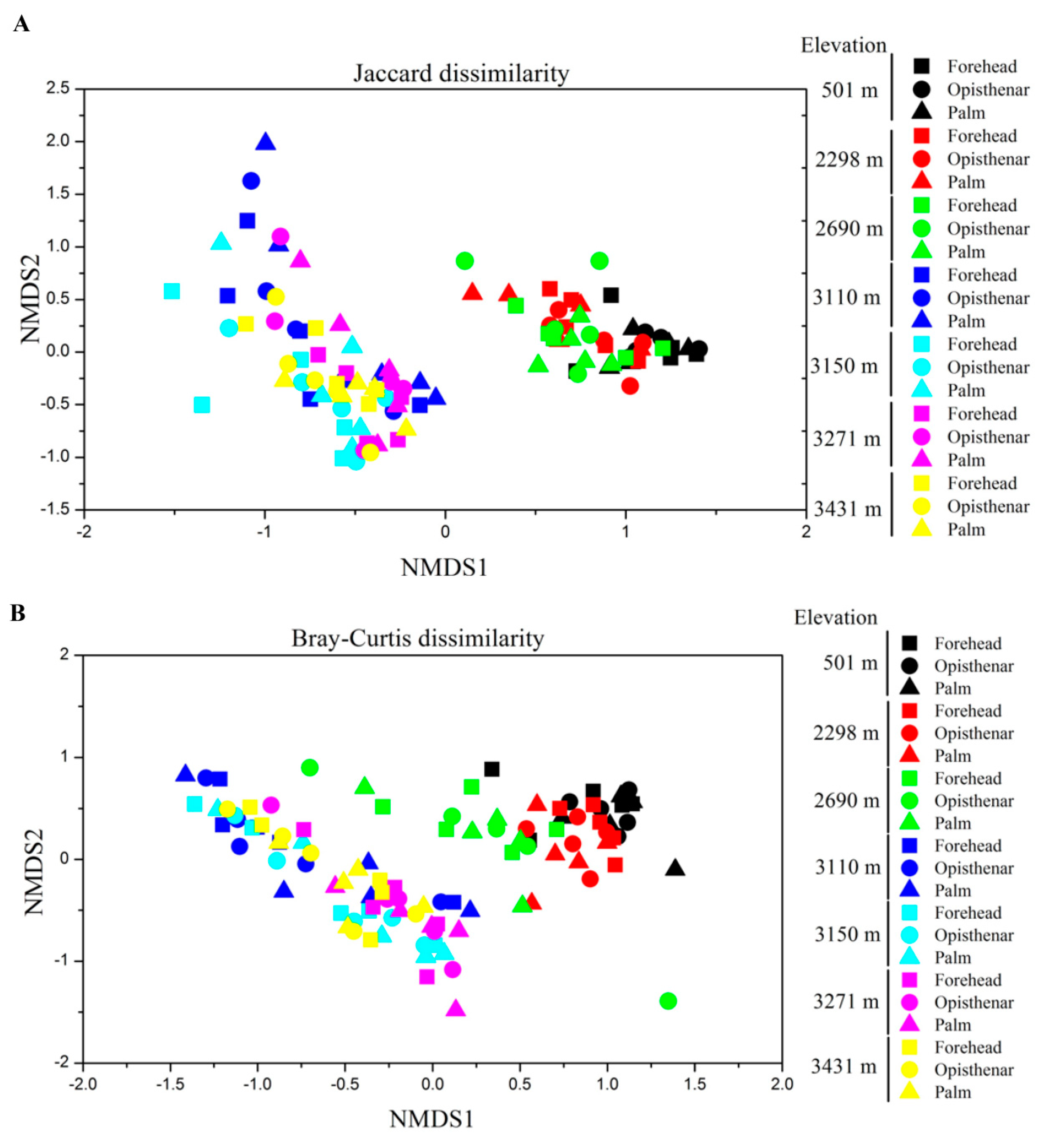
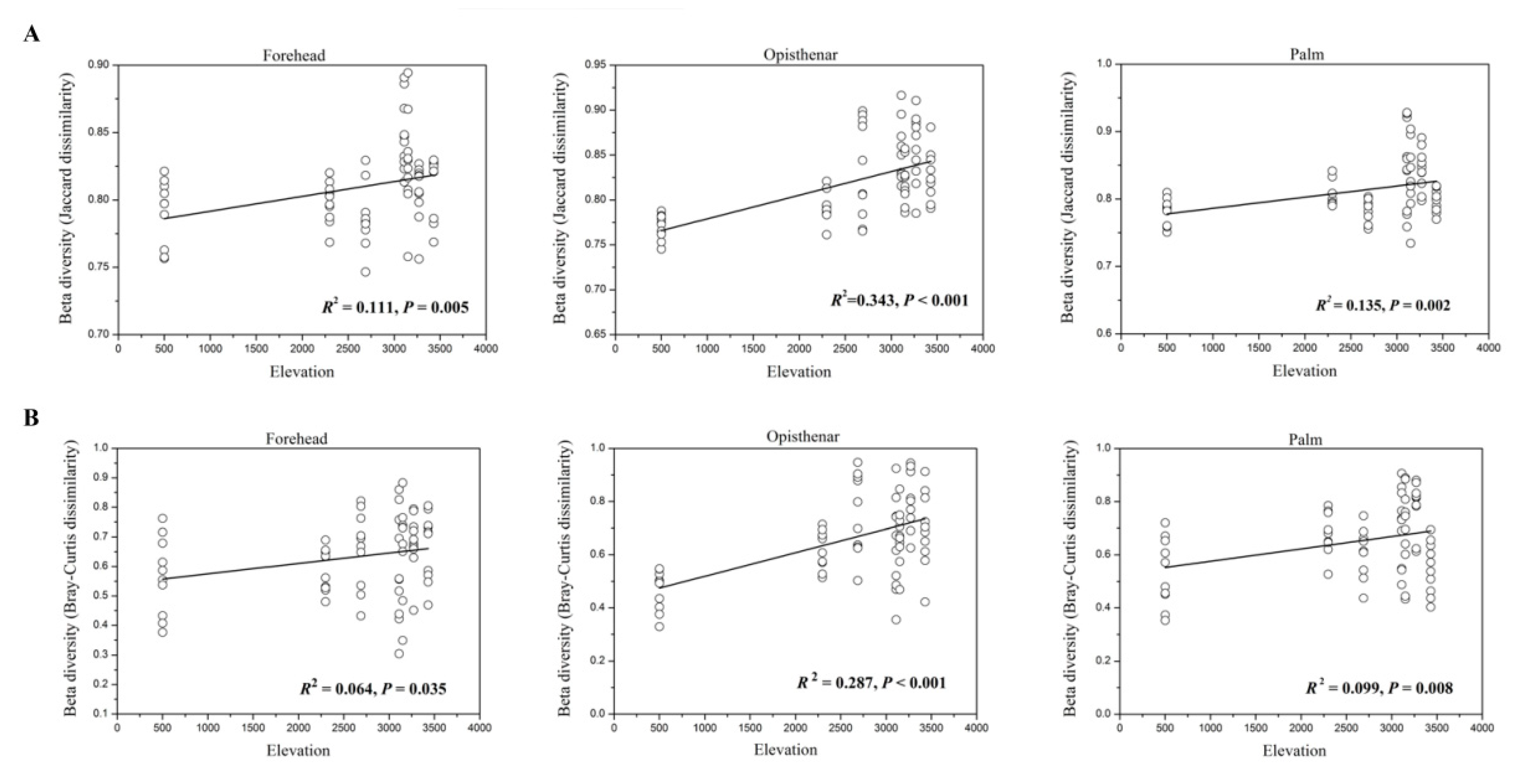
3.3. Alpha and Beta Diversity Patterns of Skin Microbiotas Along Elevational Gradients
3.4. The Differences of Predicted Gene Functions between High and Low-Elevation Skin Microbiotas
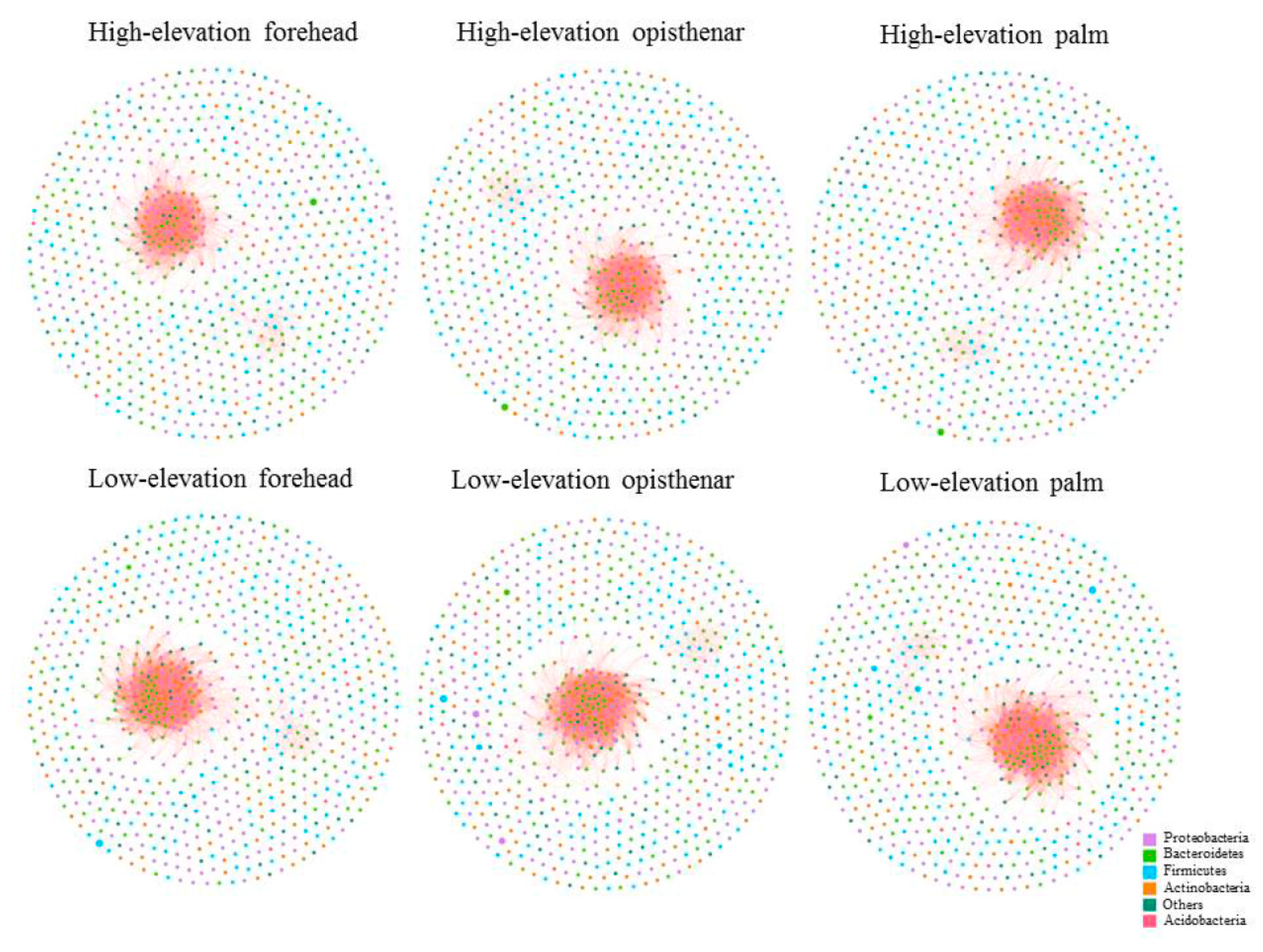
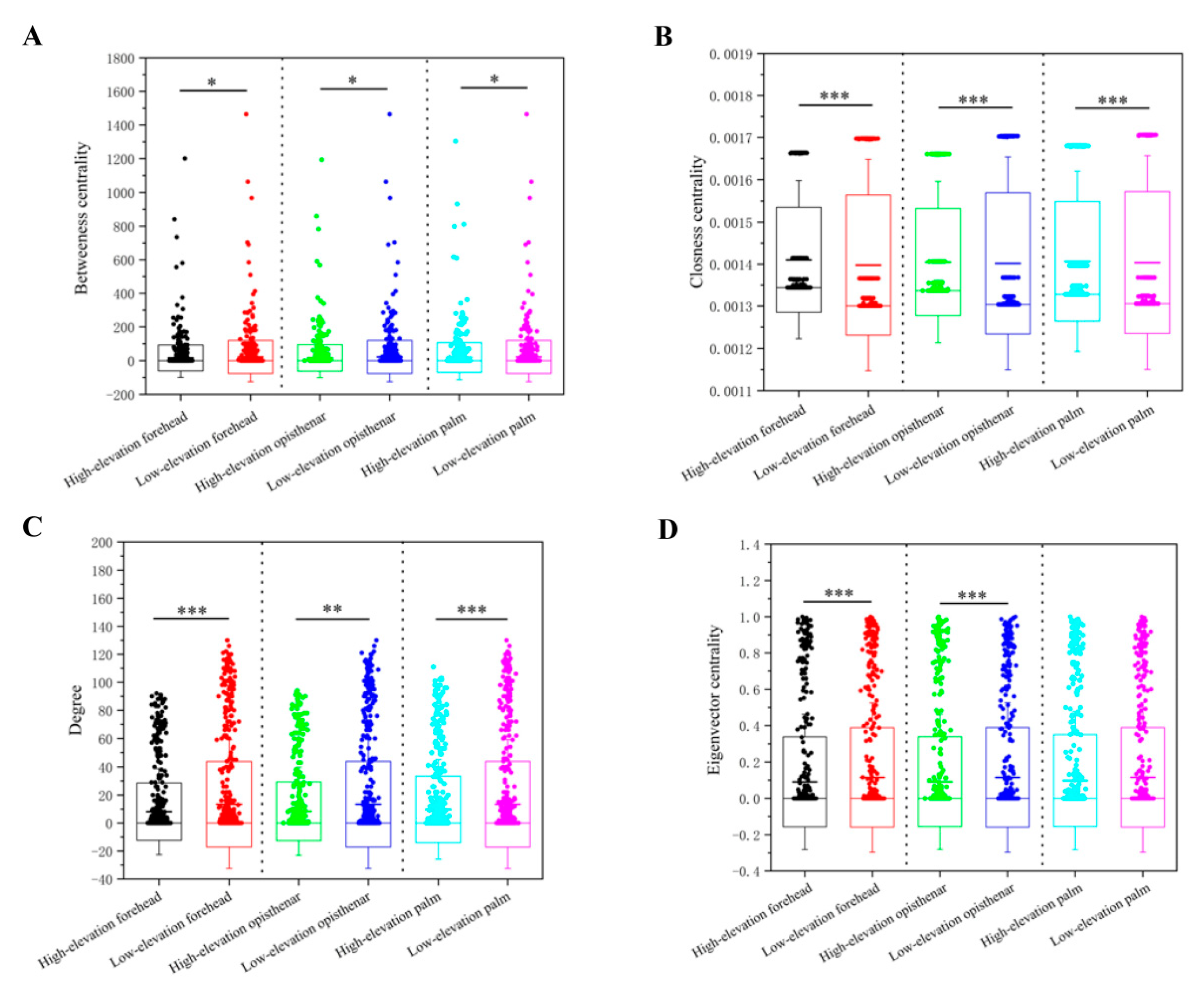
3.5. Co-Occurrence Patterns of Different Skin Subcommunities
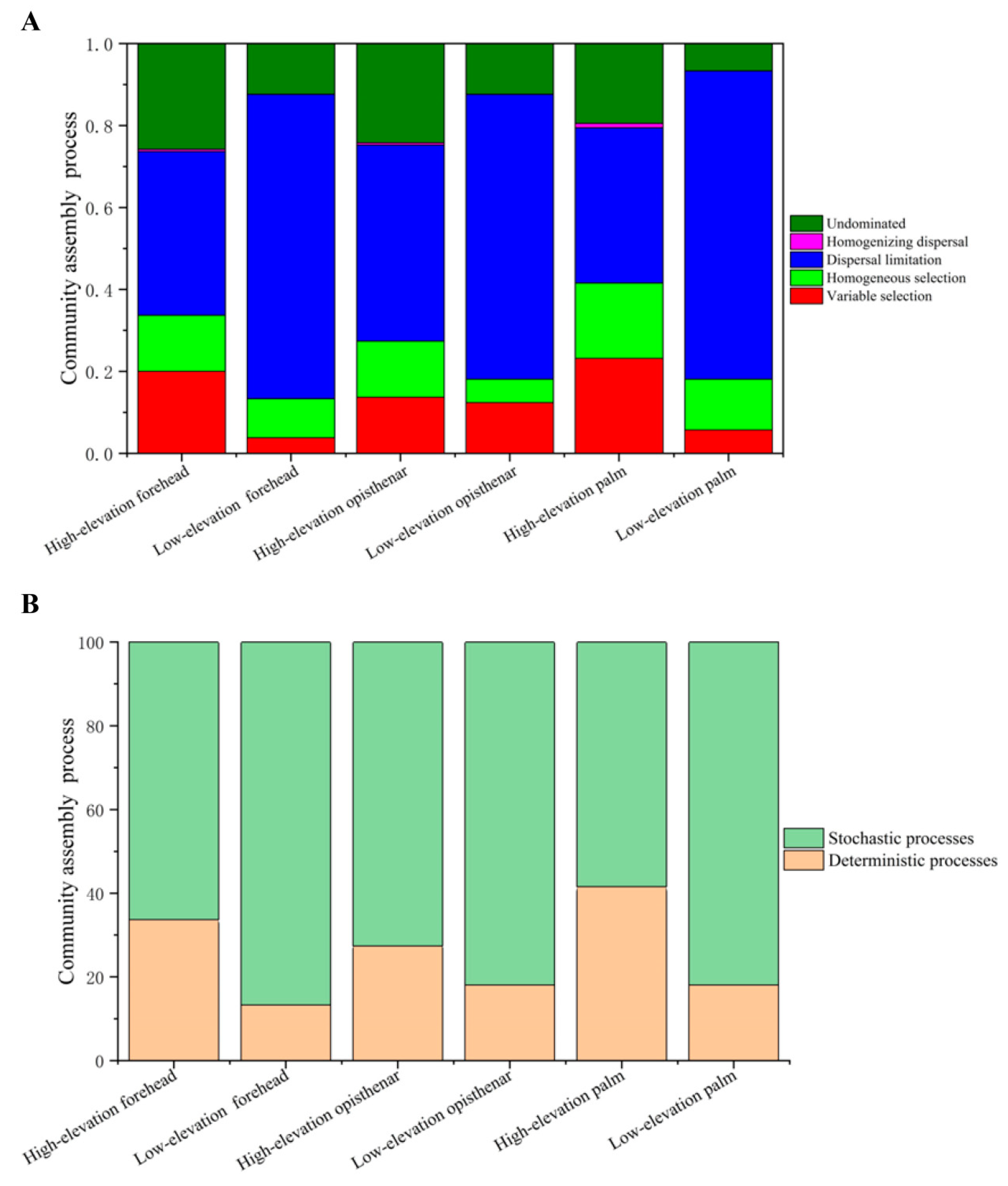
3.6. Ecological Processes Governing the Assembly of Human Skin Microbiotas
4. Discussion
4.1. Elevation-Tolerant Microbes Indicate That the Adaptation of Skin Microbiota for Extreme High-Elevation Environment
4.2. Alpha Diversity Decreases but Beta Diversity Increases for Skin Microbiota Along the Elevational Gradient
4.3. High-Elevation Skin Microbiota Networks are More Fragile Than Those at Low-Elevation Areas
4.4. Stochastic Processes Dominate the Human Skin Microbiota, but High-Altitude Skin Microbiota Harbors More Deterministic Processes
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Chen, Y.E.; Fischbach, M.A.; Belkaid, Y. Skin microbiota-host interactions. Nature 2018, 553, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Moissl-Eichinger, C.; Probst, A.J.; Birarda, G.; Auerbach, A.; Koskinen, K.; Wolf, P.; Holman, H.N. Human age and skin physiology shape diversity and abundance of Archaea on skin. Sci. Rep. 2017, 7, 4039. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Byrd, A.L.; Park, M.; Program, N.C.S.; Kong, H.H.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Tamoutounour, S. The influence of skin microorganisms on cutaneous immunity. Nat. Rev. Immunol. 2016, 16, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A. The intersection of microbiome and host at the skin interface: Genomic- and metagenomic-based insights. Genome Res. 2015, 25, 1514–1520. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Sequencing, N.I.; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Genet. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Muletz Wolz, C.R.; Yarwood, S.A.; Campbell Grant, E.H.; Fleischer, R.C.; Lips, K.R. Effects of host species and environment on the skin microbiome of Plethodontid salamanders. J. Anim. Ecol. 2018, 87, 341–353. [Google Scholar] [CrossRef]
- Perez Perez, G.I.; Gao, Z.; Jourdain, R.; Ramirez, J.; Gany, F.; Clavaud, C.; Demaude, J.; Breton, L.; Blaser, M.J. Body Site Is a More Determinant Factor than Human Population Diversity in the Healthy Skin Microbiome. PLoS ONE 2016, 11, e0151990. [Google Scholar] [CrossRef]
- SanMiguel, A.; Grice, E.A. Interactions between host factors and the skin microbiome. Cell Mol. Life Sci. 2015, 72, 1499–1515. [Google Scholar] [CrossRef]
- Grice, E.A.; Snitkin, E.S.; Yockey, L.J.; Bermudez, D.M.; Program, N.C.S.; Liechty, K.W.; Segre, J.A.; Mullikin, J.; Blakesley, R.; Young, A.; et al. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc. Natl. Acad. Sci. USA 2010, 107, 14799–14804. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chiang, H.I.; Jiang, S.B.; Nagarajan, H.; Zengler, K.; Gallo, R.L. The microbiome extends to subepidermal compartments of normal skin. Nat. Commun. 2013, 4, 1431. [Google Scholar] [CrossRef]
- Smeekens, S.P.; Huttenhower, C.; Riza, A.; van de Veerdonk, F.L.; Zeeuwen, P.L.; Schalkwijk, J.; van der Meer, J.W.; Xavier, R.J.; Netea, M.G.; Gevers, D. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. J. Innate Immun. 2014, 6, 253–262. [Google Scholar] [CrossRef]
- Wu, T.Y. Challenges of plateau hypoxic environment to humans. J. Med. Res. 2006, 35, 1–3. [Google Scholar]
- Beall, C.M. Andean, Tibetan, and Ethiopian patterns of adaptation to high-altitude hypoxia. Integr. Compar. Biol. 2006, 46, 18–24. [Google Scholar] [CrossRef]
- Jablonski, N.G.; Chaplin, G. Human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. 2), 8962–8968. [Google Scholar] [CrossRef]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; Program, N.C.S.; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef]
- Hughey, M.C.; Pena, J.A.; Reyes, R.; Medina, D.; Belden, L.K.; Burrowes, P.A. Skin bacterial microbiome of a generalist Puerto Rican frog varies along elevation and land use gradients. PeerJ 2017, 5, e3688. [Google Scholar] [CrossRef]
- Zeng, B.; Zhao, J.; Guo, W.; Zhang, S.; Hua, Y.; Tang, J.; Kong, F.; Yang, X.; Fu, L.; Liao, K.; et al. High-Altitude Living Shapes the Skin Microbiome in Humans and Pigs. Front. Microbiol. 2017, 8, 1929. [Google Scholar] [CrossRef]
- Park, T.; Kim, H.; Myeong, N.; Lee, H.; Kwack, I.; Lee, J.; Kim, B.; Sul, W.; An, S. Collapse of human scalp microbiome network in dandruff and seborrheic dermatitis. Exp. Dermatol. 2017, 26, 835–838. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Tu, B.; Kou, Y.; Li, X. Host species shapes the co-occurrence patterns rather than diversity of stomach bacterial communities in pikas. Appl. Microbiol. Biotechnol. 2017, 101, 5519–5529. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Berasategui, A.; Rui, J.; Zhang, X.; Li, C.; Xiao, Z.; Li, X. Gut region influences the diversity and interactions of bacterial communities in pikas (Ochotona curzoniae and Ochotona daurica). FEMS Microbiol. Ecol. 2017, 93, fix149. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Li, X.; Wang, G.; Lin, Q.; Qu, J. Gut Microbiota in Tibetan Herdsmen Reflects the Degree of Urbanization. Front. Microbiol. 2018, 9, 1745. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Genet. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Li, S.P.; Cadotte, M.W.; Meiners, S.J.; Pu, Z.; Fukami, T.; Jiang, L. Convergence and divergence in a long-term old-field succession: The importance of spatial scale and species abundance. Ecol. Lett. 2016, 19, 1101–1109. [Google Scholar] [CrossRef]
- Anderson, K.J. Temporal patterns in rates of community change during succession. Am. Nat. 2007, 169, 780–793. [Google Scholar] [CrossRef]
- Vellend, M. Conceptual synthesis in community ecology. Q. Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef]
- Vellend, M.; Srivastava, D.S.; Anderson, K.M.; Brown, C.D.; Jankowski, J.E.; Kleynhans, E.J.; Kraft, N.J.B.; Letaw, A.D.; Macdonald, A.A.M.; Maclean, J.E.; et al. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 2014, 123, 1420–1430. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Yao, M.; Li, J.; Zhang, S.; Wirth, S.; Cao, W.; Lin, Q.; Li, X. Pika gut may select for rare but diverse environmental bacteria. Front. Microbiol. 2016, 7, 1269. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, J.; Li, T.; Li, J.; Lin, Q.; Li, X. Pika population density is associated with composition and diversity of gut microbiota. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Beasley, D.E.; Hedenec, P.; Xiao, Z.; Zhang, S.; Li, J.; Lin, Q.; Li, X. Diet Diversity Is Associated with Beta but not Alpha Diversity of Pika Gut Microbiota. Front. Microbiol. 2016, 7, 1169. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Haas, B.; Clemente, J.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Jaccard, P. The distribution of the flora in the alpine zone. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Bray, J.; Curtis, J. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- McArdle, B.; Anderson, M. Fitting multivariate models to community data: A comment on distance-based redundancy analysis. Ecology 2001, 82, 290–297. [Google Scholar] [CrossRef]
- Benjamini, Y.; Krieger, A.; Yekutieli, D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006, 93, 491–507. [Google Scholar] [CrossRef]
- Luo, F.; Zhong, J.; Yang, Y.; Scheuermann, R.; Zhou, J. Application of random matrix theory to biological networks. Phys. Lett. 2006, 357, 420–423. [Google Scholar] [CrossRef]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Kim, J.; Myeong, N.; Kim, T.; Park, T.; Kim, E.; Choi, J.; Lee, J.; An, S.; et al. Fragile skin microbiomes in megacities are assembled by a predominantly niche-based process. Sci. Adv. 2018, 4, e1701581. [Google Scholar] [CrossRef]
- Findley, K.; Grice, E. The skin microbiome: A focus on pathogens and their association with skin disease. PLOS Pathog. 2014, 10, e1004436. [Google Scholar] [CrossRef]
- Dib, J.; Motok, J.; Zenoff, V.F.; Ordonez, O.; Farias, M.E. Occurrence of resistance to antibiotics, UV-B, and arsenic in bacteria isolated from extreme environments in high-altitude (above 4400 m) Andean wetlands. Curr. Microbiol. 2008, 56, 510–517. [Google Scholar] [CrossRef]
- Ordonez, O.F.; Flores, M.R.; Dib, J.R.; Paz, A.; Farias, M.E. Extremophile culture collection from Andean lakes: Extreme pristine environments that host a wide diversity of microorganisms with tolerance to UV radiation. Microb. Ecol. 2009, 58, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, V.H.; Pathak, G.P.; Douki, T.; Cadet, J.; Borsarelli, C.D.; Gartner, W.; Farias, M.E. Extremophilic Acinetobacter strains from high-altitude lakes in Argentinean Puna: Remarkable UV-B resistance and efficient DNA damage repair. Orig. Life Evol. Biosph. 2012, 42, 201–221. [Google Scholar] [CrossRef] [PubMed]
- Kung, S.S.; Chuang, Y.C.; Chen, C.H.; Chien, C.C. Isolation of polyhydroxyalkanoates-producing bacteria using a combination of phenotypic and genotypic approach. Lett. Appl. Microbiol. 2007, 44, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Bergogne-Berezin, E.; Towner, K.J. Acinetobacter spp. as nosocomial pathogens: Microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 1996, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Liu, W.; Cui, L.; Zhang, M.; Wang, B. Characterization and identification of a chlorine-resistant bacterium, Sphingomonas TS001, from a model drinking water distribution system. Sci. Total. Environ. 2013, 458–460, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Antoun, H.; Beauchamp, C.J.; Goussard, N.; Chabot, R.; Lalande, R. Potential of Rhizobium and Bradyrhizobium species as plant growth promoting rhizobacteria on non-legumes: Effect on radishes (Raphanus sativus L.). In Molecular Microbial Ecology of the Soil; Springer: Dordrecht, The Netherlands, 1998; pp. 57–67. [Google Scholar]
- Margalef, R. On certain unifying principles in ecology. Am. Nat. 1963, 97, 357–374. [Google Scholar] [CrossRef]
- McNaughton, S.J. Diversity and stability of ecological communities: A comment on the role of empiricism in ecology. Am. Nat. 1977, 111, 515–525. [Google Scholar] [CrossRef]
- Bannar-Martin, K.H.; Kremer, C.T.; Ernest, S.K.M.; Leibold, M.A.; Auge, H.; Chase, J.; Declerck, S.A.J.; Eisenhauer, N.; Harpole, S.; Hillebrand, H.; et al. Integrating community assembly and biodiversity to better understand ecosystem function: The Community Assembly and the Functioning of Ecosystems (CAFE) approach. Ecol. Lett. 2018, 21, 167–180. [Google Scholar] [CrossRef]
- Louca, S.; Polz, M.F.; Mazel, F.; Albright, M.B.N.; Huber, J.A.; O’Connor, M.I.; Ackermann, M.; Hahn, A.S.; Srivastava, D.S.; Crowe, S.A.; et al. Function and functional redundancy in microbial systems. Nat. Ecol. Evol. 2018, 2, 936–943. [Google Scholar] [CrossRef]
- Murillo, N.; Raoult, D. Skin microbiota: Overview and role in the skin diseases acne vulgaris and rosacea. Future Microbiol. 2013, 8, 209–222. [Google Scholar] [CrossRef]
- Mathieu, A.; Vogel, T.M.; Simonet, P. The future of skin metagenomics. Res. Microbiol. 2014, 165, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.; Caporaso, J.; Henley, J.; Rideout, J.; Domogala, D.; Chase, J.; Leff, J.; Vázquez-Baeza, Y.; Gonzalez, A.; Knight, R.; et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.; Hughey, M.C.; Becker, M.H.; Walke, J.B.; Umile, T.P.; Burzynski, E.A.; Iannetta, A.; Minbiole, K.P.C.; Belden, L.K. Variation in Metabolite Profiles of Amphibian Skin Bacterial Communities Across Elevations in the Neotropics. Microb. Ecol. 2017, 74, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Renaud, G.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Wolfsberg, T.G.; Turner, M.L.; Segre, J.A. A diversity profile of the human skin microbiota. Genome Res. 2008, 18, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Maity, C.; Ghosh, K.; Pati, B.; Mondal, K. Dynamics of predominant microbiota in the human gastrointestinal tract and change in luminal enzymes and immunoglobulin profile during high-altitude adaptation. Folia Microbiol. 2013, 58, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Hanski, I.; von Hertzen, L.; Fyhrquist, N.; Koskinen, K.; Torppa, K.; Laatikainen, T.; Karisola, P.; Auvinen, P.; Paulin, L.; Mäkelä, M.; et al. Environmental biodiversity, human microbiota, and allergy are interrelated. Proc. Natl. Acad. Sci. USA 2012, 109, 8334–8339. [Google Scholar] [CrossRef]
- Hellmann, H.; Mooney, S. Vitamin B6: A molecule for human health? Molecules 2010, 15, 442–459. [Google Scholar] [CrossRef]
- Jiao, S.; Chen, W.; Wei, G. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol. Ecol. 2017, 26, 5305–5317. [Google Scholar] [CrossRef]
- Lynch, M.D.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Genet. 2015, 13, 217–229. [Google Scholar] [CrossRef]
- Li, H.; Li, T.; Qu, J. Stochastic processes govern bacterial communities from the blood of pikas and from their arthropod vectors. FEMS Microbiol. Ecol. 2018, 94, fiy082. [Google Scholar] [CrossRef]
- Adair, K.L.; Wilson, M.; Bost, A.; Douglas, A.E. Microbial community assembly in wild populations of the fruit fly. ISME J. 2018, 12, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Stegen, J.C.; Maldonado-Gomez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Soininen, J.; He, J.; Shen, J. Phylogenetic clustering increases with elevation for microbes. Environ. Microbiol. Rep. 2012, 4, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.O.; Ackerly, D.D.; McPeek, M.A.; Donoghue, M.J. Phylogenies and Community Ecology. Annu. Rev. Ecol. Syst. 2002, 33, 475–505. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Nodes | Edges | Clustering Coefficient | Average Degree | Graph Density | Modularity |
|---|---|---|---|---|---|---|
| High-elevation forehead | 744 | 3018 | 0.7337 | 8.1129 | 0.0109 | 0.1697 |
| Low-elevation forehead | 769 | 5136 | 0.7443 | 13.3576 | 0.0174 | 0.1166 |
| High-elevation opisthenar | 748 | 3123 | 0.7269 | 8.3503 | 0.0112 | 0.1591 |
| Low-elevation opisthenar | 767 | 5130 | 0.7444 | 13.3768 | 0.0175 | 0.1130 |
| High-elevation palm | 753 | 3661 | 0.7276 | 9.7238 | 0.0129 | 0.1461 |
| Low-elevation palm | 766 | 5116 | 0.7444 | 13.3577 | 0.0175 | 0.1172 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wang, Y.; Yu, Q.; Feng, T.; Zhou, R.; Shao, L.; Qu, J.; Li, N.; Bo, T.; Zhou, H. Elevation is Associated with Human Skin Microbiomes. Microorganisms 2019, 7, 611. https://doi.org/10.3390/microorganisms7120611
Li H, Wang Y, Yu Q, Feng T, Zhou R, Shao L, Qu J, Li N, Bo T, Zhou H. Elevation is Associated with Human Skin Microbiomes. Microorganisms. 2019; 7(12):611. https://doi.org/10.3390/microorganisms7120611
Chicago/Turabian StyleLi, Huan, Yijie Wang, Qiaoling Yu, Tianshu Feng, Rui Zhou, Liye Shao, Jiapeng Qu, Nan Li, Tingbei Bo, and Huakun Zhou. 2019. "Elevation is Associated with Human Skin Microbiomes" Microorganisms 7, no. 12: 611. https://doi.org/10.3390/microorganisms7120611
APA StyleLi, H., Wang, Y., Yu, Q., Feng, T., Zhou, R., Shao, L., Qu, J., Li, N., Bo, T., & Zhou, H. (2019). Elevation is Associated with Human Skin Microbiomes. Microorganisms, 7(12), 611. https://doi.org/10.3390/microorganisms7120611