Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish and Sample Collection
2.2. DNA Extraction, PCR Amplification and Illumina HiSeq 2500 Sequencing
2.3. Quality Control and Read Assembly
2.4. OTU Cluster and Taxonomy Classification
2.5. Diversity Analysis and Functional Prediction
3. Results
3.1. Statistics of Illumina Sequencing Data and OTUs
3.2. Microbiota Structure
3.3. Diversity Differences and Potential Functions
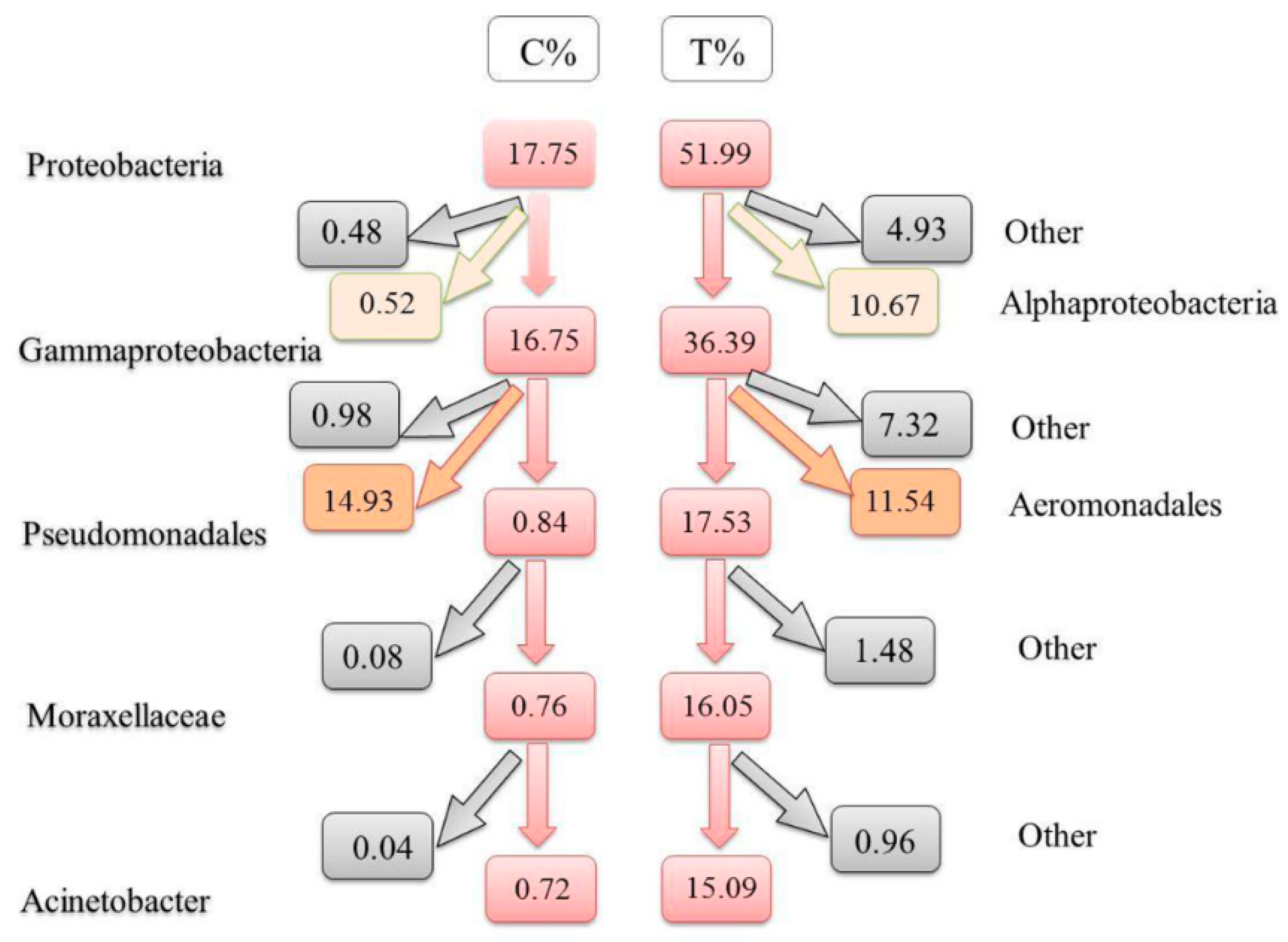
3.4. Differences at Different Taxonomic Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, E.M.; Liu, B.H.; Wang, G.J.; Yu, D.G.; Xie, J.; Xia, Y.; Gong, W.B.; Wang, H.H.; Li, Z.F.; Wei, N. Molecular cloning of type I collagen cDNA and nutritional regulation of type I collagen mRNA expression in grass carp. J. Anim. Physiol. Anim. Nutr. 2014, 98, 755–765. [Google Scholar] [CrossRef]
- Yu, E.M.; Ma, L.L.; Ji, H.; Li, Z.F.; Wang, G.J.; Xie, J.; Yu, D.G.; Kaneko, G.; Tian, J.J.; Zhang, K.; et al. Smad4-dependent regulation of type I collagen expression in the muscle of grass carp fed with faba bean. Gene 2019, 685, 32–41. [Google Scholar] [CrossRef]
- Yu, E.; Xie, J.; Wang, G.; Yu, D.; Gong, W.; Li, Z.; Wang, H.; Xia, Y.; Wei, N. Gene expression profiling of grass carp (Ctenopharyngodon idellus) and crisp grass carp. Int. J. Genom. 2014, 2014, 639687. [Google Scholar] [CrossRef]
- Yu, E.M.; Zhang, H.F.; Li, Z.F.; Wang, G.J.; Wu, H.K.; Xie, J.; Yu, D.G.; Xia, Y.; Zhang, K.; Gong, W.B. Proteomic signature of muscle fibre hyperplasia in response to faba bean intake in grass carp. Sci. Rep. 2017, 7, 45950. [Google Scholar] [CrossRef]
- Wu, S.; Ren, Y.; Peng, C.; Hao, Y.; Xiong, F.; Wang, G.; Li, W.; Zou, H.; Angert, E.R. Metatranscriptomic discovery of plant biomass-degrading capacity from grass carp intestinal microbiomes. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef]
- Lin, W.L.; Zeng, Q.X.; Zhu, Z.W.; Song, G.S. Relation between protein characteristics and Tpa texture characteristics of crisp grass carp (Ctenopharyngodon Idellus C. Et V) and grass carp (Ctenopharyngodon Idellus). J. Texture Stud. 2012, 43, 1–11. [Google Scholar] [CrossRef]
- Tian, J.J.; Ji, H.; Wang, Y.F.; Xie, J.; Wang, G.J.; Li, Z.F.; Yu, E.M.; Yu, D.G.; Zhang, K.; Gong, W.B. Lipid accumulation in grass carp (Ctenopharyngodon idellus) fed faba beans (Vicia faba L.). Fish Physiol. Biochem. 2019, 45, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F.; Sapp, J.; Tauber, A.I. A symbiotic view of life: We have never been individuals. Q. Rev. Biol. 2012, 87, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Wu, S.; Wang, G.; Angert, E.R.; Wang, W.; Li, W.; Zou, H. Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS ONE 2012, 7, e30440. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.; Wang, G.-T.; Wu, S.-G. A review of intestinal microbes in grass carp Ctenopharyngodon idellus (Valenciennes). Aquac. Res. 2017, 48, 3287–3297. [Google Scholar] [CrossRef]
- Han, S.F.; Liu, Y.C.; Zhou, Z.G.; He, S.X.; Cao, Y.A.; Shi, P.J.; Yao, B.; Ringo, E. Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aquac. Res. 2010, 42, 47–56. [Google Scholar] [CrossRef]
- Hao, Y.T.; Wu, S.G.; Xiong, F.; Tran, N.T.; Jakovlic, I.; Zou, H.; Li, W.X.; Wang, G.T. Succession and fermentation products of grass carp (Ctenopharyngodon idellus) hindgut microbiota in response to an extreme dietary shift. Front. Microbiol. 2017, 8, 1585. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, J.; Jakovlic, I.; Xiong, F.; Wu, S.; Zou, H.; Li, W.; Li, M.; Wang, G. Gut segments outweigh the diet in shaping the intestinal microbiota composition in grass carp Ctenopharyngodon Idellus. AMB Express 2019, 9, 44. [Google Scholar] [CrossRef]
- Ni, J.J.; Yan, Q.Y.; Yu, Y.H.; Zhang, T.L. Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS Microbiol. Ecol. 2014, 87, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Zhou, X.Q.; Jiang, W.D.; Wu, P.; Liu, Y.; Jiang, J.; Wang, S.W.; Kuang, S.Y.; Tang, L.; Feng, L. Effects of dietary zearalenone on oxidative stress, cell apoptosis, and tight junction in the intestine of juvenile grass carp (Ctenopharyngodon idella). Toxins 2019, 11, 333. [Google Scholar] [CrossRef]
- Li, Z.F.; Yu, E.M.; Wang, G.J.; Yu, D.G.; Zhang, K.; Gong, W.B.; Xie, J. Broad bean (Vicia faba L.) induces intestinal inflammation in grass carp (Ctenopharyngodon idellus C. et V) by increasing relative abundances of intestinal gram-negative and flagellated bacteria. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Buttigieg, P.L.; Ramette, A. A guide to statistical analysis in microbial ecology: A community-focused, living review of multivariate data analyses. FEMS Microbiol. Ecol. 2014, 90, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Ta × 4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Li, H. Preliminary study on the ecology, physiology and pathology of crisped grass carp (Ctenopharyngodon idellus C.et V). Acta Ecol. Sin. 2006, 26, 2749–2756. [Google Scholar]
- Cai, X.; Luo, L.; Xue, M.; Wu, X.; Zhan, W. Growth performance, body composition and phosphorus availability of juvenile grass carp (Ctenopharyngodon idellus) as affected by diet processing and replacement of fishmeal by detoxified castor bean meal. Aquacult. Nutr. 2005, 11, 293–299. [Google Scholar] [CrossRef]
- Gan, L.; Li, X.X.; Pan, Q.; Wu, S.L.; Feng, T.; Ye, H. Effects of replacing soybean meal with faba bean meal on growth, feed utilization and antioxidant status of juvenile grass carp, Ctenopharyngodon idella. Aquacult. Nutr. 2017, 23, 192–200. [Google Scholar] [CrossRef]
- Ingerslev, H.C.; Jorgensen, L.V.; Strube, M.L.; Larsen, N.; Dalsgaard, I.; Boye, M.; Madsen, L. The development of the gut microbiota in rainbow trout (Oncorhynchus mykiss) is affected by first feeding and diet type. Aquaculture 2014, 424, 24–34. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Munoz-Price, L.S.; Weinstein, R.A. Acinetobacter infection. New Engl. J. Med. 2008, 358, 1271–1281. [Google Scholar] [CrossRef]
- Huang, W.; Wang, G.; Yin, C.; Chen, D.; Dhand, A.; Chanza, M.; Dimitrova, N.; Fallon, J.T. Optimizing a whole-genome sequencing data processing pipeline for precision surveillance of health care-associated infections. Microorganisms 2019, 7, 388. [Google Scholar] [CrossRef]
- Rampelotto, P.H.; Sereia, A.F.R.; de Oliveira, L.F.V.; Margis, R. Exploring the hospital microbiome by high-resolution 16S rRNA profiling. Int. J. Mol. Sci. 2019, 20, 3099. [Google Scholar] [CrossRef]
- Shrestha, R.G.; Tandukar, S.; Bhandari, D.; Sherchan, S.P.; Tanaka, Y.; Sherchand, J.B.; Haramoto, E. Prevalence of Arcobacter and other pathogenic bacteria in river water in nepal. Water-Sui. 2019, 11, 1416. [Google Scholar] [CrossRef]
- Hoang Quoc, C.; Nguyen Thi Phuong, T.; Nguyen Duc, H.; Tran Le, T.; Tran Thi Thu, H.; Nguyen Tuan, S.; Phan Trong, L. Carbapenemase genes and multidrug resistance of Acinetobacter Baumannii: A cross sectional study of patients with pneumonia in southern vietnam. Antibiotics 2019, 8, 148. [Google Scholar] [CrossRef]
- Perdikouri, E.I.A.; Arvaniti, K.; Lathyris, D.; Apostolidou Kiouti, F.; Siskou, E.; Haidich, A.B.; Papandreou, C. Infections due to multidrug-resistant bacteria in oncological patients: Insights from a five-year epidemiological and clinical analysis. Microorganisms 2019, 7, 277. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Seifert, H.; Paterson, D.L. Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 2008, 21, 538–582. [Google Scholar] [CrossRef]
- Huang, H.; Dong, Y.; Yang, Z.L.; Luo, H.; Zhang, X.; Gao, F. Complete sequence of pABTJ2, a plasmid from Acinetobacter baumannii MDR-TJ, carrying many phage-like elements. Genom. Proteom. Bioinform. 2014, 12, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Reveco, F.E.; Overland, M.; Romarheim, O.H.; Mydland, L.T. Intestinal bacterial community structure differs between healthy and inflamed intestines in Atlantic salmon (Salmo salar L.). Aquaculture 2014, 420, 262–269. [Google Scholar] [CrossRef]
- Yao, J.; Chang, L.; Yuan, L.; Duan, Z. Nutrition status and small intestinal bacterial overgrowth in patients with virus-related cirrhosis. Asia Pac. J. Clin. Nutr. 2016, 25, 283–291. [Google Scholar]
- Bures, J.; Cyrany, J.; Kohoutova, D.; Forstl, M.; Rejchrt, S.; Kvetina, J.; Vorisek, V.; Kopacova, M. Small intestinal bacterial overgrowth syndrome. World J. Gastroenterol. 2010, 16, 2978–2990. [Google Scholar] [CrossRef]
- Yamini, D.; Pimentel, M. Irritable bowel syndrome and small intestinal bacterial overgrowth. J. Clin. Gastroenterol. 2010, 44, 672–675. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Distance | R | p Value | Significant# |
|---|---|---|---|---|
| TF/TM | Bray-Curtis | −0.12 | 0.854 | |
| TF/TH | Bray-Curtis | 0.34 | 0.049 | * |
| TM/TH | Bray-Curtis | 0.25 | 0.073 | |
| CF/CM | Bray-Curtis | −0.04 | 0.731 | |
| CF/CH | Bray-Curtis | 0.30 | 0.018 | * |
| CM/CH | Bray-Curtis | 0.28 | 0.017 | * |
| TF/CF | Bray-Curtis | 1.00 | 0.002 | ** |
| TM/CM | Bray-Curtis | 0.98 | 0.005 | ** |
| TH/CH | Bray-Curtis | 0.62 | 0.003 | ** |
| T/C | Bray-Curtis | 0.87 | 0.001 | ** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Lin, K.-t.; Gan, L.; Sun, J.-j.; Guo, C.-j.; Liu, L.; Huang, X.-d. Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study. Microorganisms 2019, 7, 465. https://doi.org/10.3390/microorganisms7100465
Zhou L, Lin K-t, Gan L, Sun J-j, Guo C-j, Liu L, Huang X-d. Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study. Microorganisms. 2019; 7(10):465. https://doi.org/10.3390/microorganisms7100465
Chicago/Turabian StyleZhou, Lei, Ke-tao Lin, Lian Gan, Ji-jia Sun, Chang-jun Guo, Li Liu, and Xian-de Huang. 2019. "Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study" Microorganisms 7, no. 10: 465. https://doi.org/10.3390/microorganisms7100465
APA StyleZhou, L., Lin, K.-t., Gan, L., Sun, J.-j., Guo, C.-j., Liu, L., & Huang, X.-d. (2019). Intestinal Microbiota of Grass Carp Fed Faba Beans: A Comparative Study. Microorganisms, 7(10), 465. https://doi.org/10.3390/microorganisms7100465